
WHAT IS SICKLE CELL DISEASE?
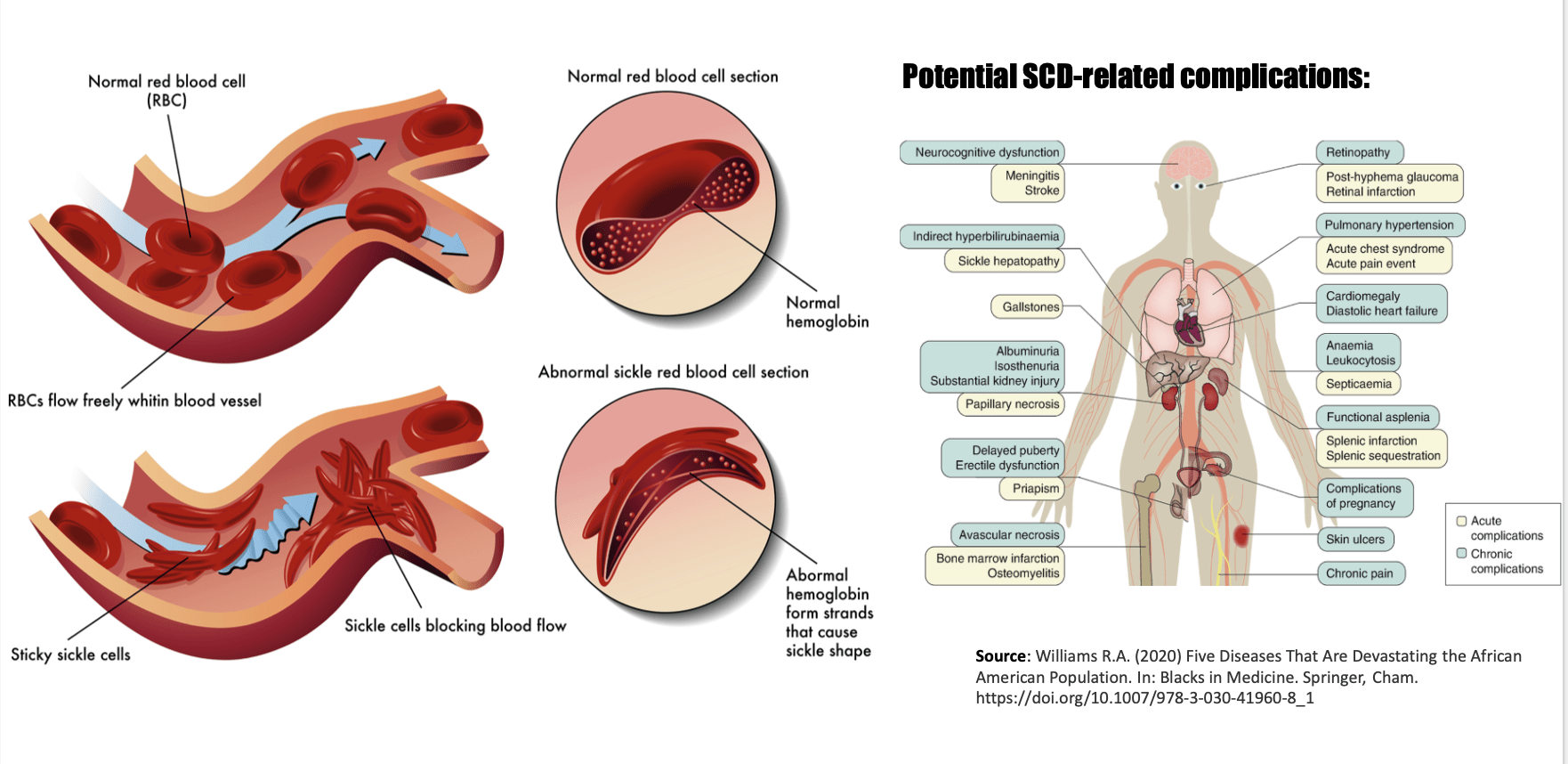
Sickle cell disease (SCD) is a life-threatening, inherited blood disorder which causes red blood cells to become sickle-shaped through the presence of an abnormal variant of hemoglobin. This variant, known as HbS, is created by a single mutation in the beta subunits of healthy HbA (adult hemoglobin): a Glutamate at position 6 of the primary sequence is substituted by a Valine. This change in structure on the surface of the protein from a positively charged amino acid to a non-polar, hydrophobic amino acid causes affected blood cells to shrivel up and aggregate to one another, thereby inhibiting circulation. This subsequently leads to a large number of bodily complications which may include an increased risk for infection, organ damage, stroke, and even death.
While SCD symptoms may begin in patients around 6 months of age, it’s nearly impossible to diagnose them at that time due to the fact that infants are unable to vocalize their pain. This phenomenon, known as the Pain Crisis, is a large contributing factor to the staggering mortality rate in Sub-Saharan regions of the world.
GLOBAL IMPACT OF SCD
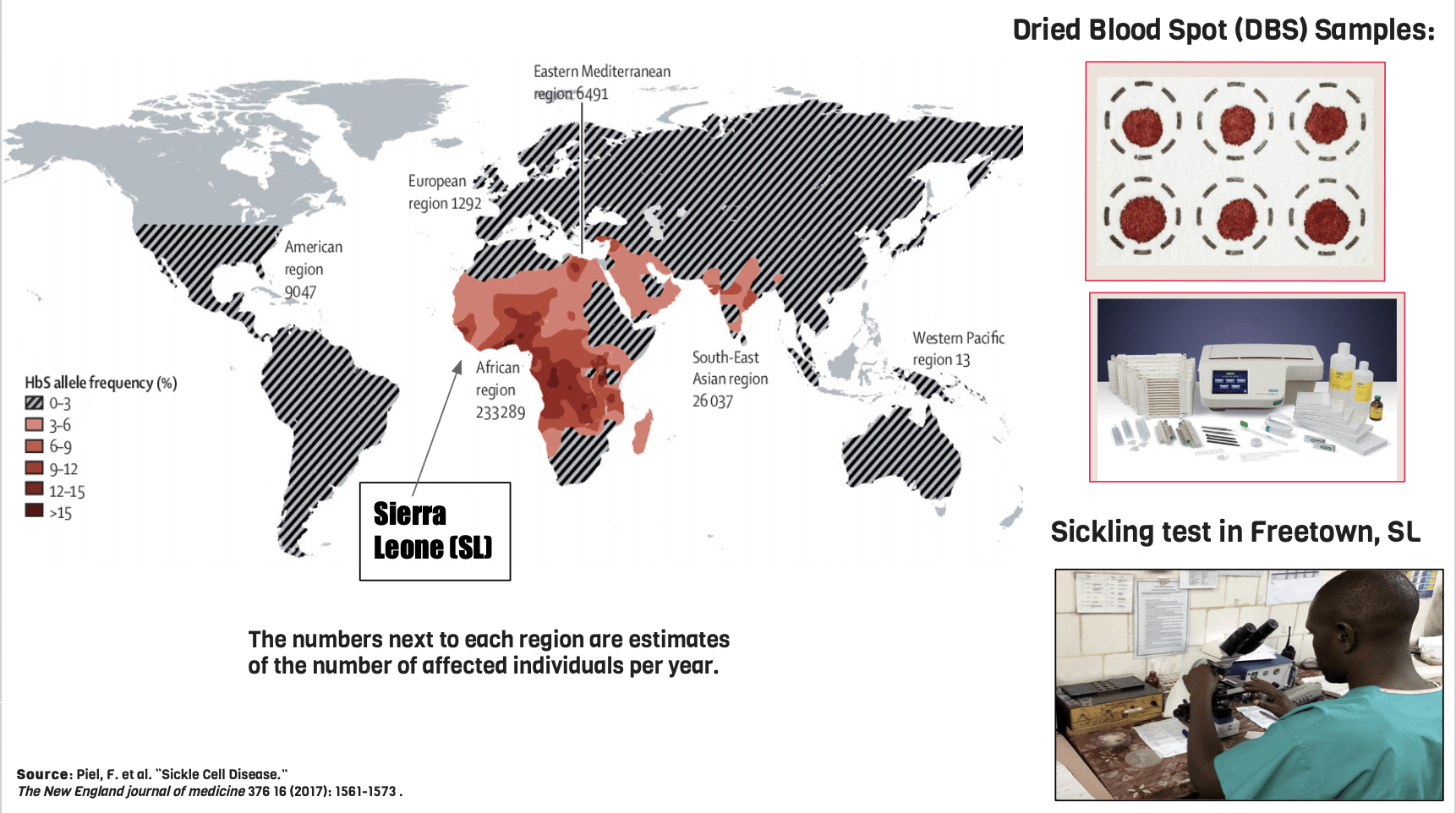
SCD has worldwide impact. In 2019, there was an estimate of 300k babies born each year with SCD, and over 230k of these individuals are in Sub-Saharan Africa. 50-90% of these individuals in this region die before the age of 5. While SCD has some prevalence in higher income countries, such as the US, these regions have been able to significantly increase life expectancy by adopting universal screening programs to catch the illness early. However, LMICs are unable to implement these solutions due to lack of resources.
The current programs that have been implemented in LMICs include the collection of dried blood spot (DBS) samples and the Sickling Test. DBS samples are collected from newborns and then sent to centralized labs for analysis. The problem with these initiatives relates to their time-consuming and expensive nature. Costs of transportation to external labs and the use of the gold standard (hemoglobin electrophoresis and high-performance liquid chromatography) to carry out the test are too high for many low-to-middle income countries to implement sustainably. That’s why many countries, including Sierra Leone, rely on the Sickling Test. The Sickling Test is a qualitative experiment which involves taking a drop of blood, adding adding a deoxygenating reagent, and then observing the response of the blood cells. This test operates under the assumption that a crescent shape will form for patients with SCD. The issue here is that it is too difficult to differentiate between individuals who carry the sickling trait and patients who have sickle cell disease.
In short, the use of either DBS samples or the Sickling Test raises issues in accuracy or cost burden. Our team hopes to come in as a solution, with our device optimized to cost under five dollars while still being designed to achieve the same level of diagnostic accuracy as a comparable $12-15 electrophoresis screening test to be run.