Introduction.
Most experts agree that stress is the critical factor that precipitates major depression. Those prone to depression have greater susceptibility, but presumably everyone has some threshold at which depression would occur.
Over the years there have been at least 5 different hypotheses of what goes wrong in the brain between experiencing stress and the development of depression. However, the explanations are not mutually exclusive, with each hypothesis helping to understand different aspects of the disorder. While each hypothesis has utility, none provides a comprehensive understanding. It is also possible that that major depression is not a single disorder but rather multiple disorders with overlapping symptoms. There is still much we don’t understand.
The Monoamine Hypothesis. The monoamine hypothesis is the oldest hypothesis and derives from the first effective antidepressant drugs that work by boosting monoamine neurotransmitter concentrations in the brain. According to this idea, stress suppresses monoamine neurotransmission which in turn leads to depression.
There are 5 different monoamine neurotransmitters in the brain (serotonin, norepinephrine, epinephrine, dopamine and histamine), however since the monoamine antidepressants target mainly serotonin and norepinephrine, these 2 neurotransmitters have received the most attention. However, there is some evidence that dopamine might play a lesser role.
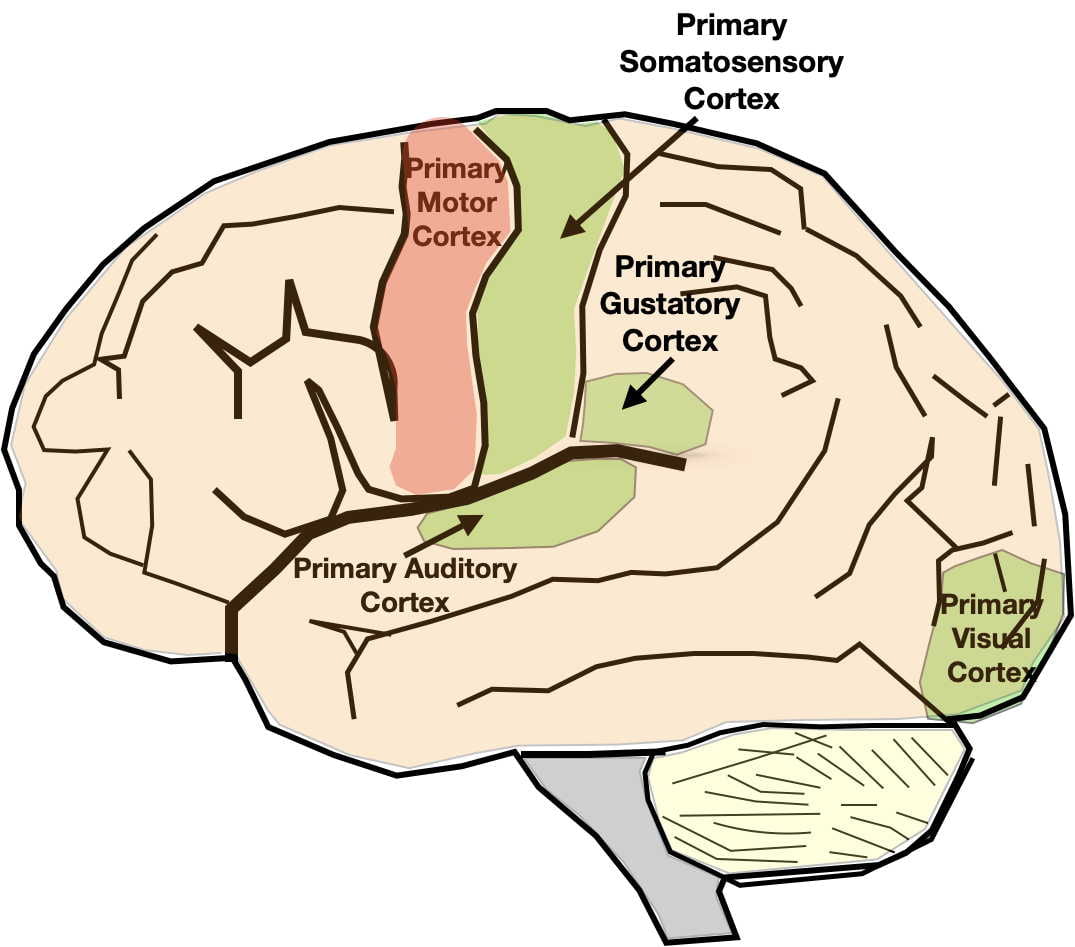
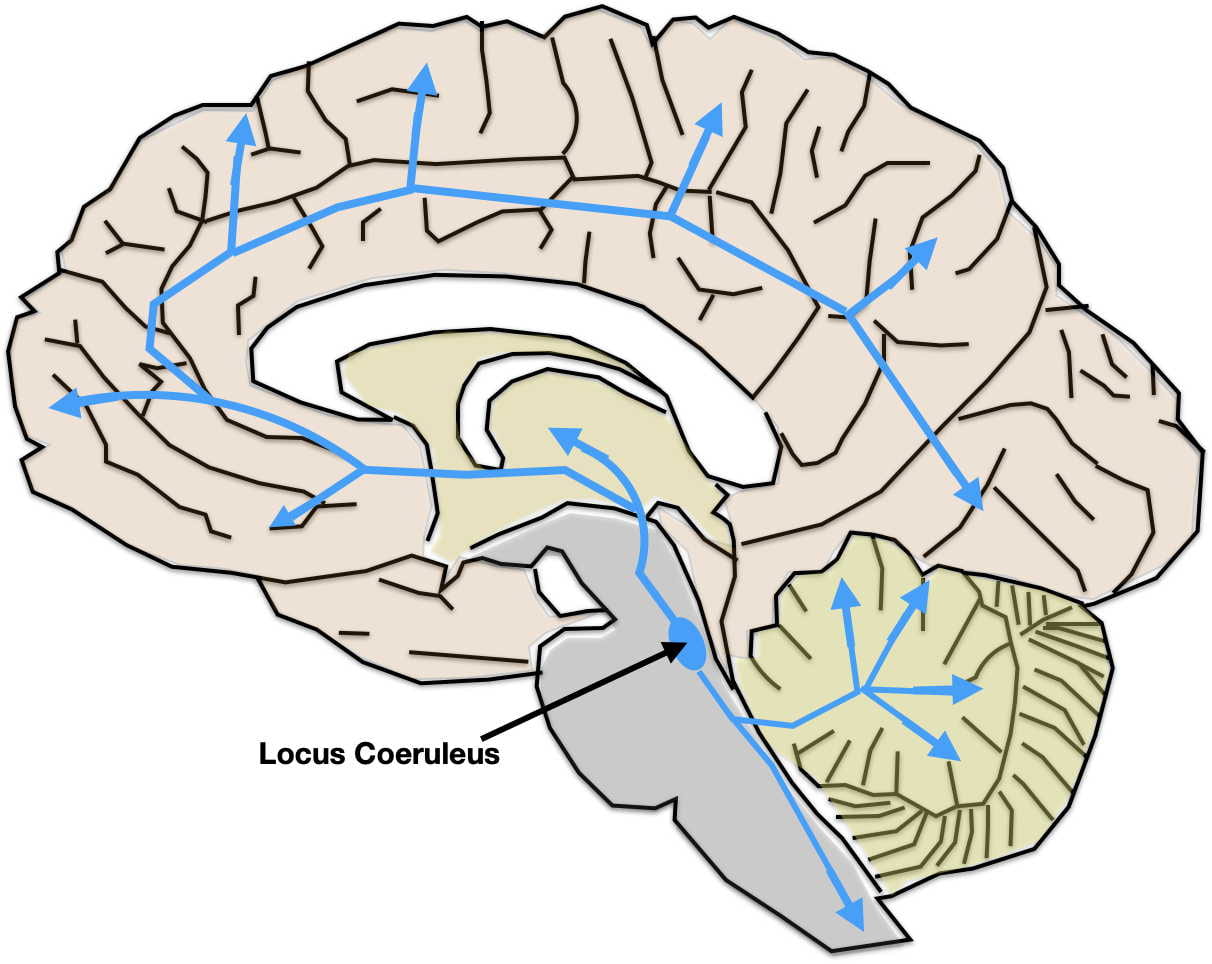
As illustrated at the end of the previous post, the serotonin and norepinephrine pathways in the brain possess the anatomical and functional characteristics one might expect of systems underlying depression. Although their neuron cell bodies are in the brainstem, their axons terminate throughout the brain, including all the areas involved in depression.
While monoamine neurotransmitters are released across synapses, their role in depression is more likely due to a type of non-synaptic release called “volume transmission.” In volume transmission the monoamine is released into the extracellular fluid outside of synapses where it can affect receptors on multiple nearby neurons. In some cases, the neurotransmitters may be targeted to their receptors by the flow of the interstitial fluid. The receptors that respond are also thought to be largely outside of synapses. Neurotransmitter release by volume transmission is thought to provide a “monoamine bath” in the area of release whose varying concentration modulates or fine tunes local neuronal activity. At the same time, some minimal average concentration is thought necessary to maintain baseline functioning. More about this in the next post.
In the original formulation of this hypothesis, depression was thought to be caused by serotonin and norepinephrine concentrations dropping below some minimal level. However, it was initially puzzling that antidepressants restore brain monoamine concentrations within hours, while recovery takes much longer, typically from three to six weeks.
However, a now well-known principle of neurotransmitter communication is that when neurotransmitter concentrations drop, responding neurons often compensate by upregulating (i.e. increasing) their receptors. This compensation allows continued functioning despite low neurotransmitter concentrations. However, this compensation works only up to a point. Eventually communication breaks down and, in the case of monoamine neurotransmitters, depression results. Consistent with this explanation, the time course to restore normal receptor concentrations with monoamine antidepressants more closely matches the time course for depression recovery. So the thinking is that you must have a proper balance between monoamines and monoamine receptors for normal functioning
However, a problem with the monoamine hypothesis is that monoamine antidepressants do not work for all depressed individuals. Approximately 50% of patients do not respond upon their first try, and after increasing dosage or changing to an alternative antidepressant, about 30% remain non-responsive. Another issue concerns the relative importance of the different monoamines. We’re not exactly sure how the relative concentrations of serotonin, norepinephrine, and dopamine are related to depression onset.
It is perhaps relevant to the monoamine hypothesis that psychedelic drugs that interact with a type of serotonin receptor also seem to provide relief from depression. More on this in a later post.
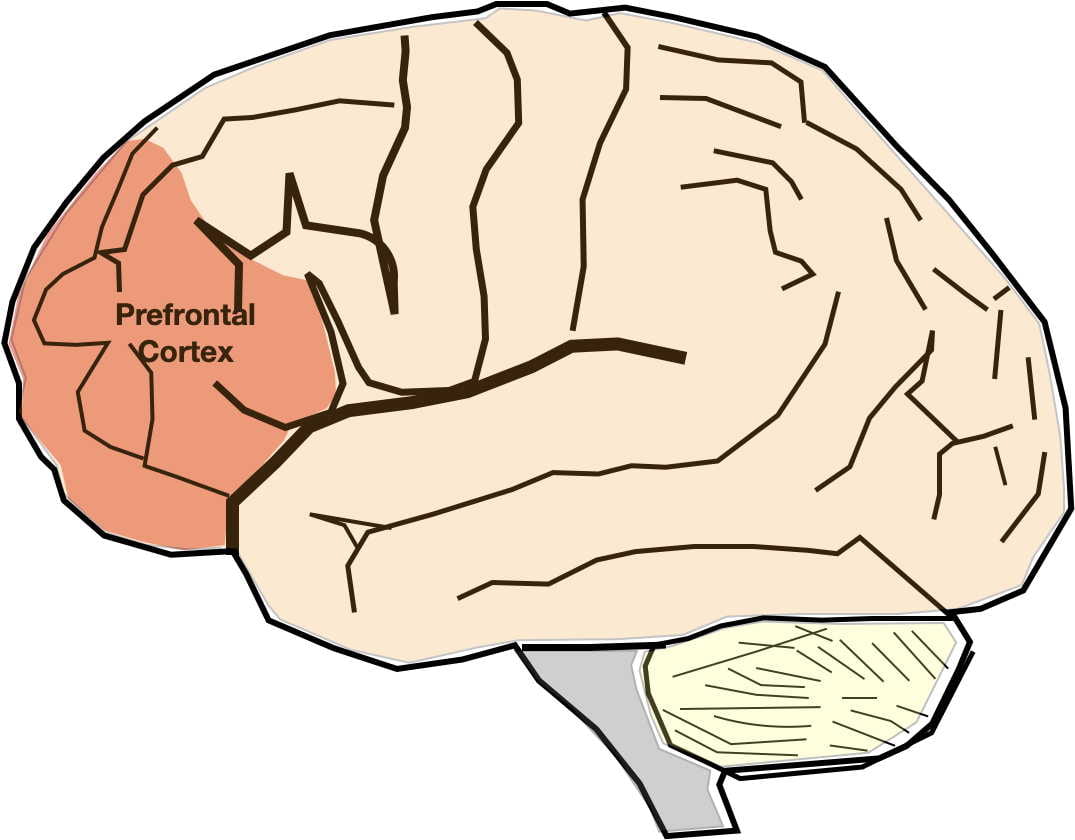
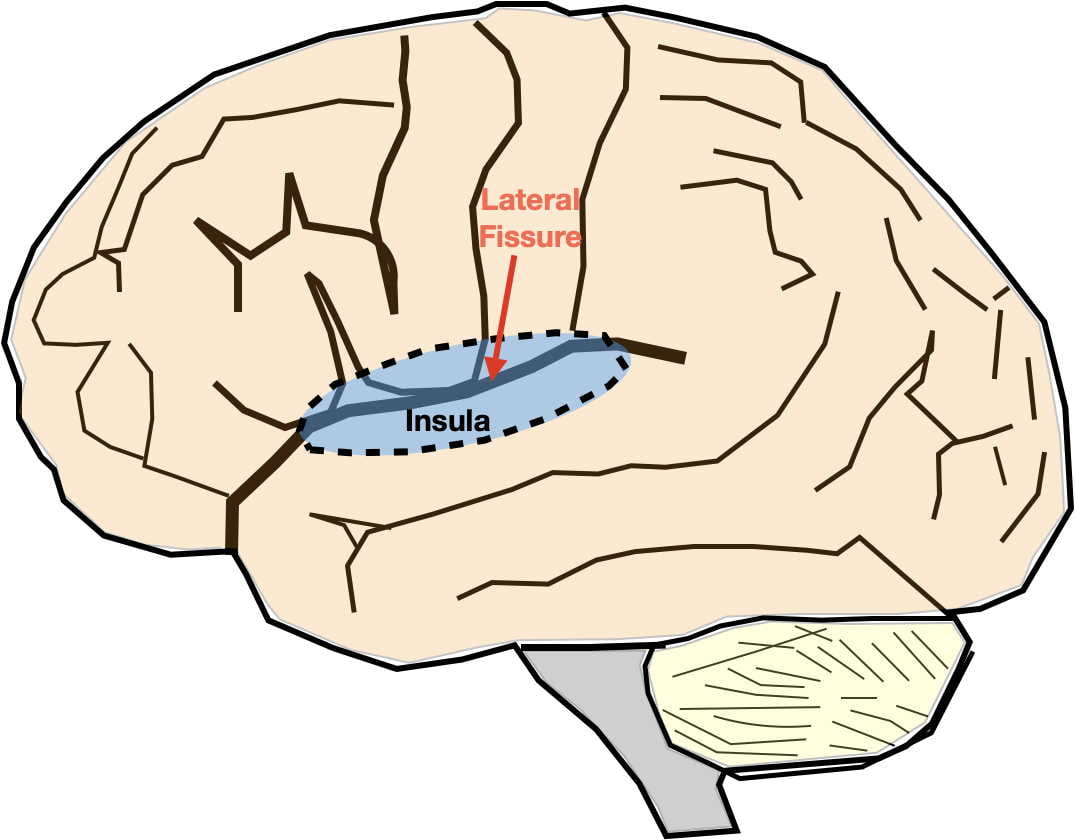
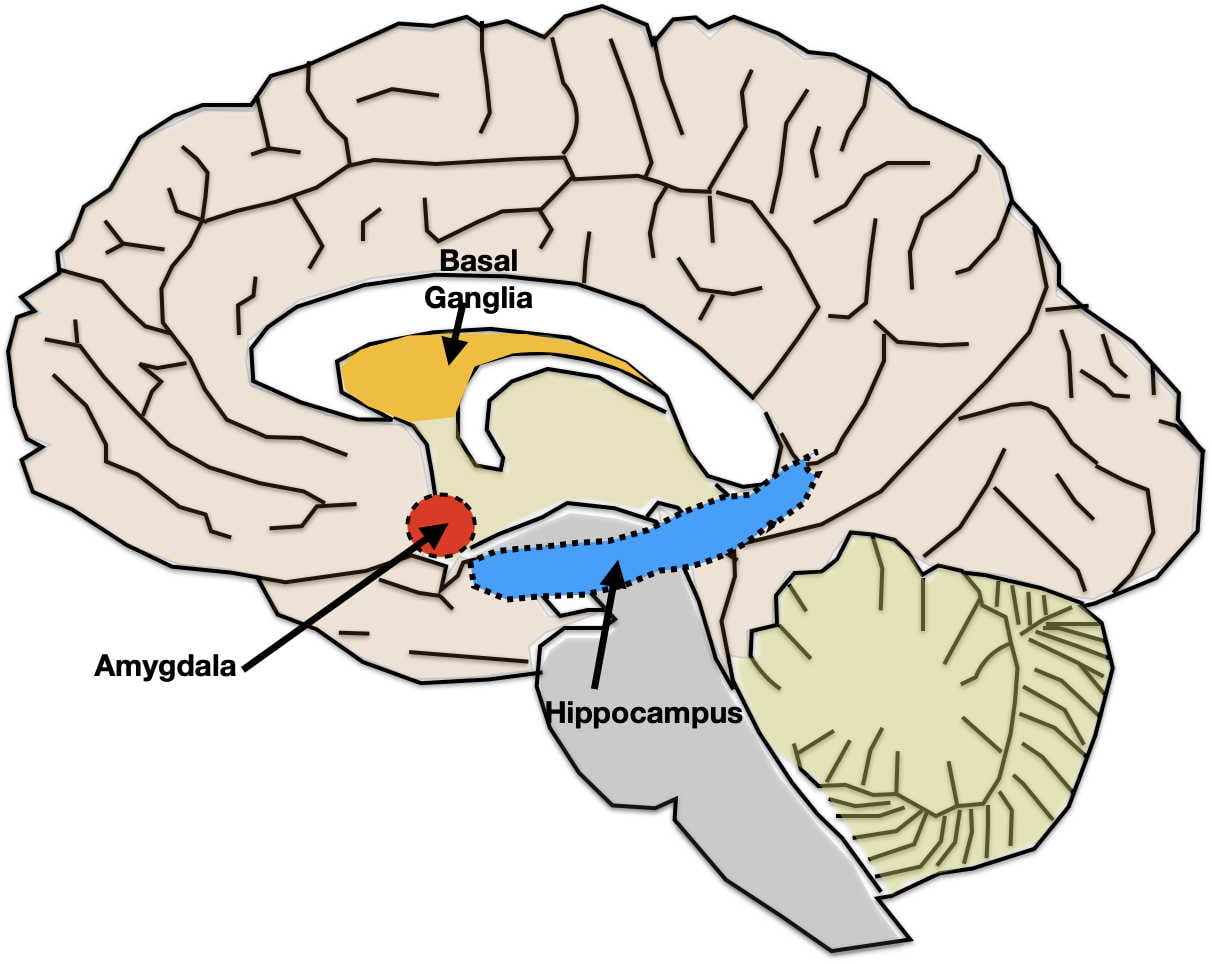
Hippocampal Neurodegeneration Hypothesis. According to this hypothesis, depression is precipitated by stress causing hippocampal degeneration. Brain scans have demonstrated that the hippocampal volume can shrink by as much as 20-30% in severely depressed individuals. Hippocampal degeneration is then thought to lead to dysfunction in the other brain areas with which it interconnects such as the prefrontal cortex, insula, anterior cingulate cortex, and amygdala. Conversely, successful depression treatment has been observed to increase hippocampal volume and restore functioning in associated areas.
As the model would predict, in stressed rodents with hippocampal degeneration, treatment with a monoamine-related antidepressant increases hippocampal volume. In some respects, the hippocampus neurodegeneration hypothesis might be viewed as an extension of the monoamine hypothesis by specifying the brain area where monoamine dysfunction initiates depressive symptomology.
The hippocampus is certainly best known for its essential role in helping the cerebral cortex to consolidate short-term memories into long-term memories and for its contributions to Alzheimer’s Disease when it degenerates. Hippocampal dysfunction in depression almost certainly also accounts for the memory deficits seen in depressed individuals as well.
One question not answered to everyone’s satisfaction concerns the process that causes the hippocampus to shrink. The hippocampus is thought by many scientists to be the only brain area in adult humans capable of neurogenesis (the birth of new neurons). The argument is that stress turns off hippocampal neurogenesis. When neurogenesis stops, dying neurons are not replaced, and the hippocampus shrinks. While the weight of evidence would seem supportive there are non-supportive findings as well. So whether shrinkage is due to a lack of neurogenesis or some other factor is currently debated.
Circadian Rhythm Abnormality. According to this idea, depression is hypothesized to be caused by the improper functioning of the biological clocks that control our 24-hour circadian rhythms.
All body processes show circadian rhythms with metabolic peaks and troughs at appropriate times of day to optimize body functioning. Although every cell of the body has its own clock, they are all synched to a “master clock” in the hypothalamus of the brain. Surprisingly, human circadian clocks typically have around a 25-hour (rather than 24-hour) periodicity (it can vary a bit from person to person). So, in order for our biological rhythms to be synched to the appropriate time of day, we must set our clocks back about an hour each day. The resetting normally occurs when we wake up each morning by “zeitgebers” (German for time givers), the most important of which is the morning light.
Circadian rhythm abnormalities are often seen in major depression, both in timing and in amplitude. The most obvious expression is in the occurrence of sleep disorders. Sleep issues can occur as amplitude problems where the person gets too little or too much sleep, and/or as temporal problems with difficulties in falling asleep or waking up at the proper time. Physiological variables such as body temperature and hormone secretion often show parallel disturbances. Successful treatment for depression typically reduces these circadian abnormalities.
Interestingly, both serotonin and norepinephrine show pronounced circadian fluctuations in brain concentrations. Both are highest when awake and physically active, drop during wakeful inactivity, drop even further upon entry into sleep, and, in the case of serotonin, drop to virtually zero during Rapid Eye Movement (REM) Sleep.
Several behavioral treatments for depression are consistent with our understanding of these circadian variations in monoamine concentration. For example, sleep deprivation, or even selective REM-sleep deprivation can improve mood in some depressed individuals. These treatments are thought to work by reducing time with low brain monoamine concentrations. Alternatively, boosting day-time monoamine concentrations through exercise can also treat mild cases of depression. Further implicating circadian rhythms, certain gene alleles that code for clock functioning have been linked to higher depression risk.
For people with Seasonal Affective Disorder (SAD), the circadian problems are even more pronounced. During the short day lengths of winter, individuals with this disorder are unable to reset their clocks each day. As a result, circadian clocks begin to “free run”, resulting in physiology losing its synchronization to the time of day. Depression is thought to be precipitated by the resulting dysfunction. While some SAD suffers respond to antidepressants, a more desirable solution is to expose them to an intense bank of lights after awakening each morning to reset their clocks. These individuals are also advised to spend as much time as possible outside during the daylight hours to further insure proper synchronization of their circadian clocks.
Depression Set-Point Abnormality. According to this idea, each individual has a unique set-point for the amount of stress necessary to precipitate depression. Predisposed individuals are thought to have low set-points while depression-resistant individuals have higher set points. For reasons that aren’t altogether clear, electrically stimulating the brain is thought to raise the set-point and, in so doing, treat depression. Several different stimulation methods have some degree of effectiveness. For a variety of reasons, these methods are mainly “last-resort” therapies when other approaches don’t work. These procedures are sometimes used for other brain disorders as well.
Electroconvulsive shock therapy is the most effective non-drug treatment available for patients unresponsive to antidepressants or psychotherapy. Electrodes placed on the skull to affect both hemispheres, or just one hemisphere, transmit high-voltage, low-amperage electricity through the cortex, resulting in an epileptic-like seizure. The procedure is performed in a hospital settings under sedation with individuals normally receiving 8-12 treatments over a period of 3 or so weeks. Some patients subsequently undergo maintenance treatments around one month apart to prevent relapse.
Memory issues and headaches can occur immediately after treatment, but typically clear up with time. While the treatment may sound terrible (particularly if you saw the movie “One Flew Over the Cuckoo’s Nest”), this treatment has been highly refined over the years and is considered safe. Research indicates that this treatment has an overall success rate of 90% in some studies, making it more effective than monoamine antidepressants. In some cases, electroconvulsive shock has also provided lifesaving relief from suicidal ideology.
Repetitive transcranial magnetic stimulation (rTMS) is a less invasive alternative to electroshock therapy, particularly for patients with less treatment-resistant depression and is most often used to augment antidepressant therapy and/or psychotherapy. Neurons in the prefrontal lobe are repeatedly stimulated by a powerful alternating magnetic field from magnets placed outside the skull. Like electroshock therapy, the treatment can be either bilateral or unilateral. The patient typically receives treatments for 5 days per week over a period of 4-6 weeks. Unlike electroshock therapy this procedure can be performed in a doctor’s office or clinic and does not cause epileptic seizures or require sedation. When used alone, it is less effective than electroshock therapy but approximately as effective as antidepressant drugs.
Yet another way of stimulating the brain is by stimulating the vagus nerve, a cranial nerve that services much of the body. A stimulating device is surgically implanted under the skin of the chest with a wire connecting to the left vagus nerve where it passes through the neck on its way to the brain. By repeatedly stimulating the vagus nerve, various parts of the brain are also stimulated. However, it may take several months of treatment to see effects on depression and is less consistently effective than either electroshock or transcranial magnetic stimulation.
Although more commonly used for Parkinson’s Disease, deep-brain stimulation (DBS) has been used to treat depression and is reasonably well tolerated. Electrodes are surgically implanted into the brain to deliver electrical stimulation directly to brain tissue. A number of brain sites have resulted in therapeutic effects. Despite discrete electrode placements, this approach does not allow for inferences about therapeutic brain sites since the stimulation quickly spreads to areas outside the stimulation site. Because of its invasiveness, this treatment is rarely used for depression.
Glutamate hypothesis. According to this hypothesis, the overrelease of glutamic acid, a brain neurotransmitter, causes the brain dysfunctions that underly depression. This hypothesis is based upon the finding that ketamine, an anesthetic drug that acts as a glutamic acid antagonist, has antidepressant properties.
Glutamic Acid (GA), also referred to as glutamate, is the major excitatory neurotransmitter of the brain and is central to the functioning of all the forebrain structures implicated in depression. Paradoxically, too much GA neurotransmission causes “excitotoxicity” (which poisons the brain). GA neurons are normally kept in check by neurons that release Gamma Amino Butyric Acid (GABA), the principle inhibitory neurotransmitter in the brain. Abnormal Levels of both GA and GABA have been observed both in depressed humans and in animal models of depression.
Support for the importance of GA neurons was first provided in 2010. In the initial study, depressed individuals, previously determined to be unresponsive to the therapeutic effects of monoamine antidepressants, received a single subanesthetic injection of ketamine. 80% showed symptomatic relief! Moreover, the therapeutic effects occurred within hours (rather than the weeks typical for the monoamine antidepressants). Unfortunately, the effects typically wear off within a week or so; however, some patients have had successful maintenance therapy for over a year, with 2 to 7-day dosing intervals.
Despite the seeming success of ketamine, it is not an ideal antidepressant. Because it is a DEA-controlled drug with short-term debilitating effects and potentially addictive properties, self-administration is illegal. The treatment by a physician is also costly, and, since it is not FDA-approved for this purpose, insurance typically does not cover costs. Currently, it is used mainly for patients unresponsive to monoamine antidepressants.
Although a new form of ketamine (esketamine) recently received FDA approval for treating depression, making it eligible for insurance coverage, it still must be administered by a physician and possesses the same side effects. A drug that provides ketamine’s benefits without its downsides, would certainly be highly desirable. There are currently attempts to develop such drugs. More about that in a later post.
To complicate matters further, GA neurons and monoamine neurons are reciprocally interconnected. The GA neurons of forebrain areas that contribute to depression are modulated by extracellular monoamine concentrations, while GA synaptic input into serotonin, norepinephrine, and dopamine neurons undoubtedly influences monoamine secretion as well. Ketamine’s antidepressant action also restores normal concentrations of brain monoamines.
Still a lot of pieces of the depression puzzle that don’t yet fit together!




{kind=link}