Introduction.
A previous post presents 5 different hypotheses for what goes wrong in the brain to cause depression. The hypotheses are not mutually exclusive, each addressing a different aspect of this complicated disorder. This post re-examines one of the more recent hypotheses, the glutamatergic hypothesis, which provides the most comprehensive understanding of depression and its treatment. According to this hypothesis, depression is ultimately caused by the malfunctioning of brain neurons that release glutamic acid as their neurotransmitter. (Hereafter glutamic acid will be referred to as glutamate, its ionic form in the brain).
The glutamatergic hypothesis arose when ketamine, a glutamate antagonist, was found in 2000, not only to treat depression quicker than first-line SSRIs (within hours vs 3-5 weeks), but was also effective in many “treatment-resistant” patients that don’t respond to SSRI’s. At the time, this discovery was viewed as the biggest depression-treatment breakthrough in 50 years.
This post examines how maladaptive glutamate activity in the brain might lead to depression, the causes of maladaptive glutamate activity, and how some new findings might lead to better antidepressant drugs.
Background on Glutamate.
Although glutamate was not recognized as a neurotransmitter until the early 1980’s, we now know that it is the most common, and most important, neurotransmitter in the brain. Glutamate is the brain’s principal excitatory neurotransmitter with its large myelinated neurons providing the primary basis for transmitting and encoding complex information. In fact, Sanacora et al. (2012) refer to the brain as “largely a glutamatergic excitatory machine.” In the human cortex and limbic system, around 80% of the neurons and 85% of the synapses are excitatory with the vast majority being glutamatergic. This glutamatergic circuitry is central to the complex neural processing underlying learning, memory, cognition, emotion, and consciousness. The other 20% of neurons and 15% of synapses are inhibitory and serve mainly to modulate the excitatory circuitry. Given its anatomical ubiquity and functional importance, it makes sense that dysfunctional glutamate signaling might play a central role in depressive illness.
In what follows I present a model for how the glutamatergic hypothesis of depression might work. Since research on humans to support many aspects of this hypothesis is lacking, some interpretive leaps between research on humans and non-human animals are necessary to make everything fit together. So, a healthy skepticism is warranted.
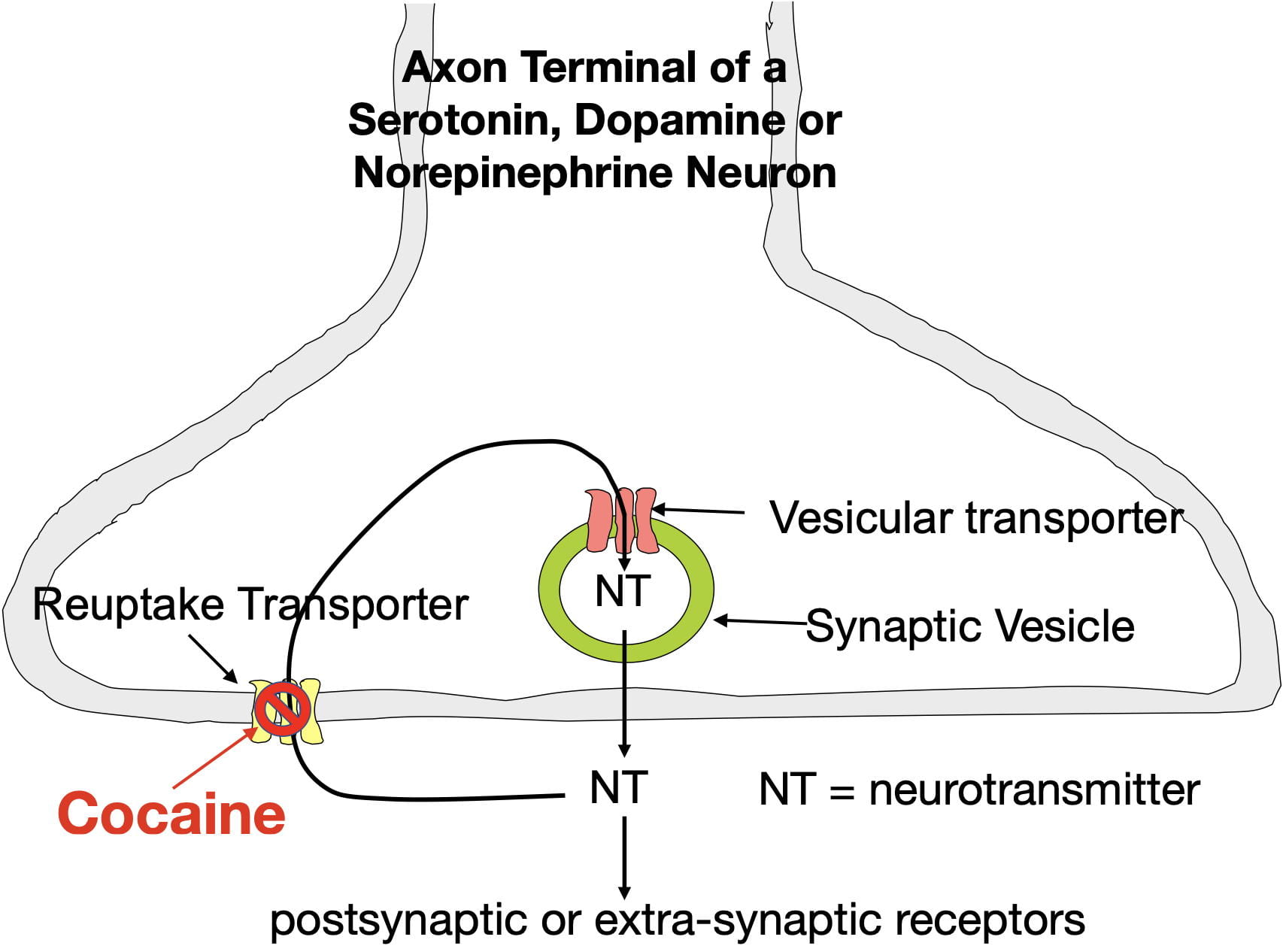
Although glutamate is present in our diet (both as a component of protein and as a free amino acid) dietary sources do not contribute much to the glutamate that neurons use as a neurotransmitter. This is because the blood brain barrier is largely impermeable to glutamate (and glutamine, its precursor). Instead, the brain’s glutamate depends mainly on the synthesis of glutamine from glucose inside astrocytes, a type of glia cell in the brain. The glutamine can then be transferred to glutamate neurons for conversion to glutamate. Figure 1 below illustrates the synthesis of glutamate, its packaging into vesicles, its release into the synapse, its removal from the synapse, and its recycling.
Glutamate is synthesized inside glutamate neuron terminals. To get into the synaptic vesicle, glutamate is actively transported across the vesicle membrane by vesicular glutamate transporters (VGLUTs), proteins embedded in the vesicle membrane. In contrast to most other small neurotransmitters which possess only a single type of vesicular transporter, glutamate has three (VGLUTs 1, 2, & 3). Once inside the vesicle, the glutamate is released into the synapse by the vesicle membrane fusing with the presynaptic membrane.
Following release, glutamate can bind to a variety of postsynaptic and presynaptic glutamate receptors in a “lock and key” fashion in which glutamate’s 3-dimensional shape fits precisely into the receptor site. Similar to other neurotransmitters, glutamate possesses high receptor specificity and low receptor affinity. This means that while glutamate binding is specific for glutamate receptors, binding does not last very long. After binding, glutamate quickly falls off its receptors and is rapidly removed from the synapse to terminate its activity. If not removed, glutamate would continue to rebind its receptors, which, as explained below, can have negative consequences.
Glutamate also has a more complex system for its synaptic removal than other neurotransmitters. Large neurotransmitters (such as peptides) often simply diffuse out of the synapse, while many small neurotransmitters are removed by a single type of transporter that moves them back into the presynaptic terminal (a process called reuptake). As seen in figure 1, glutamate. like other small neurotransmitters, undergoes reuptake by a single type of transporter (EEAT3). However, reuptake accounts for only a small fraction of synaptic removal. In fact, the more numerous EEAT1 and EEAT2 transporters in astrocytic endfeet are responsible for around 80% of the glutamate removal (Gulyaeva, 2017; Haroon et al., 2017). There are also 2 other transporters (EEAT3 and EEAT4) that move glutamate into the postsynaptic neuron. Much of the removed glutamate can eventually be reused (Mother Nature is a great recycler!).
So why does “Mother Nature” expend extra effort on glutamate synaptic-vesicle packaging and synaptic removal? One reason is to optimize the success of glutamate’s critical role in information transmission. In order to be released into the synapse, glutamate must first be packaged into synaptic vesicles. Multiple vesicular transporters help to insure this process. Once released into the synapse by action potentials entering the terminal, information is encoded in the magnitude and frequency of “pulses” of glutamate release. However, for a glutamate pulse to be detected by postsynaptic receptors, the previous pulse must be quickly removed (synaptic concentrations can change dramatically in milliseconds!). The many synaptic transporters optimize the success of this critical process.
A second important reason for the complex glutamate infrastructure is because glutamate differs from most neurotransmitters in being “excitotoxic!” Paradoxically, extended high, extracellular concentrations in the brain can “poison” and, in extreme cases, kill neurons! According to the glutamatergic hypothesis, glutamate excitotoxicity is the initial process that leads to depression (and can play a role in other neurological disabilities as well).
Fortunately there are tightly regulated features that normally protect against glutamate excitotoxicity. One feature, already described, involves the multiple transporters contributing to synaptic removal. As mentioned in other posts, the more important a biological process is to survival, the more likely it is to have redundancy.
Also protective are other types of neurons whose neurotransmitters inhibit glutamate activity. Most important in the brain are the Gamma Amino Butyric Acid (GABA) neurons, with GABA being the brain’s principle inhibitory neurotransmitter. (Paradoxically glutamate is also a precursor in the synthesis of GABA inside GABA terminals.) Like glutamate neurons, GABA neurons are found throughout the brain and often synapse onto glutamate neurons. Their major role is to tamp down glutamate activity to prevent glutamate neurons from becoming too active. Glycine, plays the same role in the spinal cord where it replaces GABA as the principal inhibitory neurotransmitter. So a balance between GABA/glycine inhibition and glutamate excitation is essential to keeping the glutamate circuitry in check.
Other neurotransmitters also participate. Depending upon who you talk to, there may be over 100 “secondary” neurotransmitters in the brain. The neurons releasing these neurotransmitters typically have 2 features in common: 1) they are regional in distribution and 2) almost all are inhibitory. These neurons help GABA neurons further refine glutamate activity in particular brain areas.
Glutamate synapses also have another unusual feature. Unlike most other synapses, glutamate synapses are completely enclosed by the endfeet of astrocytes (see figure 1). In addition to clearing glutamate from the synapse, these endfeet have the added benefit of preventing glutamate from leaking out of the synapse and inappropriately stimulating neighboring neurons and glia.
Despite all the safeguards, given the right circumstances, the glutamate system can still become overactive. This overactivity could be due to a variety of factors including excessive synaptic release, low synaptic removal, or alterations in postsynaptic receptor sensitivity. Regardless of cause, depression is thought to be one of the outcomes.
Mechanism of excitotoxicity.
So how does glutamate release cause excitotoxicity? The “big picture” is that glutamate receptor binding opens calcium channels in postsynaptic neurons. When open, calcium, more prevalent in the extracellular fluid, flows to the inside of the postsynaptic neuron. This entry activates intracellular enzymes, boosting various aspects of cellular metabolism.
While some boosting is essential to normal functioning, like so many things in life, too much of a “good thing” is bad. Excessive overstimulation can exhaust the neuron’s energy resources and also produce metabolic waste faster than it can be removed. Over time this excitation impairs the neuron’s functioning and, in extreme cases, can cause neuron death. Impaired glutamate functioning also adversely affects other types of neurons and glia cells which further contributes to depression symptomology. As the depression worsens, the individual shows signs of lessened glutamate activity (Moriguchi et al, 2019) and brain damage (Zhang et al., 2017). Fortunately, some of the changes appear reversible if the depression is effectively treated.
Now that you have the big picture, it’s time to get a little nerdy. If you’d rather not, feel free to skip the next section.
Molecular events involved in excitotoxicity.
There are actually 4 classes of glutamate receptors: 1) AMPA 2) Kainate, 3) NMDA, and 4) Metabotropic (also referred to as G-protein coupled receptors). AMPA stands for α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, Kainate stands for Kainic acid, and NMDA stands for N-Methyl-D-aspartic acid. For obvious reasons, I’ll stick to abbreviations.
AMPA, Kainate, and NMDA receptors are structurally very similar, each composed of 4 proteins that join together to form a protein complex stretching across the postsynaptic membrane. Each protein complex was named for the glutamate analog (AMPA, NMDA, or kainite) that selectively binds only its own receptor complex and not the other complexes. In fact this selective binding provided the initial evidence that these 3 receptor complexes are actually different.
AMPA, Kainate, and NMDA receptors are also referred to as ionotropic receptors because the protein complex contains not only a glutamate binding site but also an ion channel through the postsynaptic membrane (see figure 2 below). When glutamate binds its binding site, the channel almost instantaneously opens, and when glutamate falls off its binding site, the channel almost instantaneously closes. The opening and closing of the ion channel is caused by slight changes in the shape of the protein complex. To further complicate the situation, some of the contributing protein subunits have multiple isoforms (i.e. slightly different amino acid sequences) resulting in “subtypes” for each type of receptor. The subtypes are sometimes differentially expressed in different parts of the brain (and in different individuals). These subtypes may contribute to differences in susceptibility to excitotoxicity in different brain areas, and in different individuals.
In addition there are 8 different metabotropic glutamate receptors found in postsynaptic membranes and as autoreceptors in presynaptic membranes (autoreceptors provide feedback to the presynaptic terminal allowing it to adjust its release). For the metabotropic receptors, the receptor and the ion channel are separate membrane proteins in which several steps intervene between glutamate binding and opening/closing the channel. Consequently metabotropic ion channels are somewhat slower to open and, once open, remain open somewhat longer.
A glutamate synapse typically contains a mixture of ionotropic and metabotropic receptors, with receptors found both postsynaptically and presynaptically. There’s definitely a lot going on in glutamate synapses!
Two postsynaptic glutamate receptors that can cause excitoxicity are the NMDA and Kainate receptors. However, since the conventional perspective has been that ketamine exerts its antidepressant effect through binding the NMDA receptor, most of the scientific attention has been on it. The NMDA receptor is also much more widely expressed in the brain and more is known about it. (In fact, the NMDA receptor may be the brain’s most studied neurotransmitter receptor since its functioning is also central to how the brain “rewires” itself to encode memories, a story for another day.)
In general, ion channels can be opened in one of two ways: 1) chemically gated: by neurotransmitters or neurohormones binding a receptor site or 2) electrically gated: by changes in membrane voltage (either depolarization or hyperpolarization depending upon the channel). However, the NMDA ion channel is unique in being both electrically and chemically gated! This reminds me of an old Saturday Night Live skit where a salesman was selling a product that was both a floor wax and a mouthwash. 😀 Like that product, the NMDA receptor/channel complex is two products for the price of one!
Before explaining the steps involved in opening the NMDA ion channel, you need to know that when a neuron membrane is just hanging out, doing nothing, it exhibits what’s called a resting potential. At the resting potential, the inside of the membrane is negatively charged with respect to the outside because of the unequal distribution of 4 ions dissolved in the fluids on the two sides of the membrane. Sodium (Na+) and chloride (Cl-) are more common outside the neuron in the extracellular fluid, while potassium (K+) and protein anions (A-) are more common in the cytoplasm. Sodium, potassium and chloride ions all have their own channels that, when open, allow them to pass through the cell membrane. However, the protein anions, which are synthesized inside the neuron, are much larger in size and are trapped inside the neuron. At the resting potential the ion channels are closed, and with slightly more negative ions inside the neuron and slightly more positive ions outside, there is a small voltage difference across the membrane (-70 mV on the inside with respect to the outside).
There are also other ions in the brain’s fluids, but they occur in much lower concentrations and, as a result, do not contribute much to the resting potential. For example, one of these ions, calcium (Ca++) is more prevalent outside the neuron. Despite its low concentration, calcium entry into the postsynaptic neuron is the critical event that can cause excitotoxicity and depression (according to the glutamatergic hypothesis).
Figure 2 illustrates the numbered steps involved in glutamate opening the NMDA calcium ion channel that lead to excitoxicity. For reference, the relative distributions of the relevant ions are seen in the boxes.
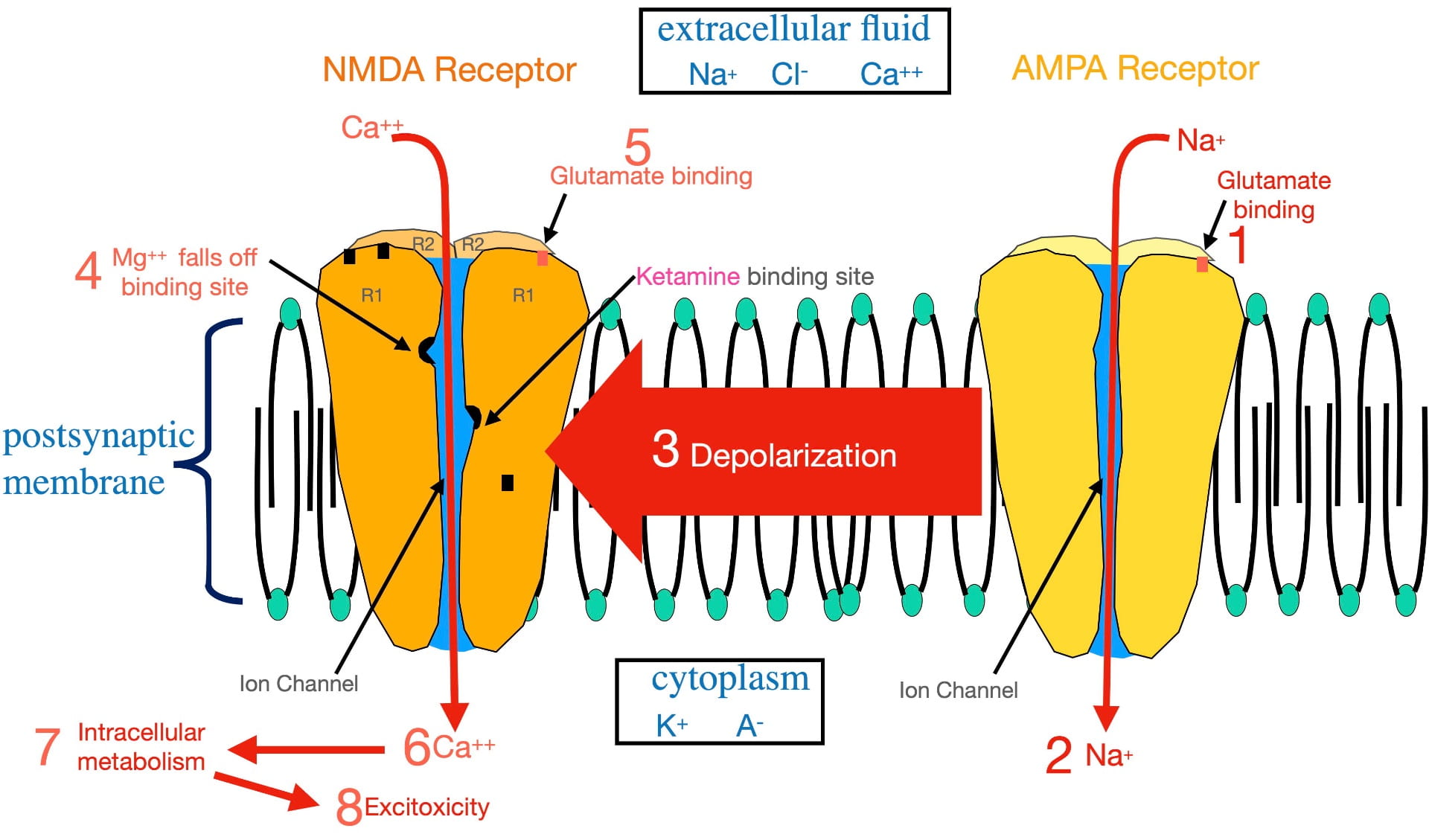
Glutamate cannot open the NMDA ion channel when the postsynaptic membrane is at the resting potential. In order to open this ion channel, the membrane must first become depolarized. This depolarization is accomplished by glutamate binding AMPA receptors present in the same postsynaptic membrane (step 1). The AMPA complex contains an ion channel selective for sodium. Glutamate binding opens the AMPA sodium channel allowing sodium to flow to the inside (step 2), depolarizing the membrane.
This depolarization then radiates out along the membrane as a graded potential (step 3). (A graded potential is one that loses voltage as it travels.) However, over the microscopically short distances involved, when the graded potential reaches an NMDA receptor, it is still sufficiently depolarized to dislodge a magnesium ion bound inside the NMDA ion channel (step 4) that is physically blocking ion flow. Once removed, glutamate binding (step 5) can open the ion channel.
Once opened, calcium passes through the NMDA channel to the inside of the postsynaptic neuron (step 6). As described earlier, this entry stimulates intracellular metabolism (step 7). This activation of intracellular metabolism is further augmented by the release of calcium from endoplasmic reticulum storage inside the neuron. If there is chronic overstimulation, excitotoxicity (step 8) can result.
To complicate matters, the NMDA “calcium” channel is not very selective. When open, not only calcium, but also sodium, flows to the inside; while potassium flows to the outside. However, the movement of these ions electrically cancel each other out, resulting in little effect on membrane voltage. The important outcome, illustrated in figure 2, is the calcium entry! (Not exactly the way I would design this channel, but it obviously does the job).
As mentioned earlier, the strongest evidence supporting the glutamatergic hypothesis is the effective treatment of depression with subanesthetic doses of ketamine. Ketamine’s antidepressant effect has historically been thought to occur by ketamine attaching to a binding site inside the ion channel separate from the magnesium site (shown in figure 2). By binding the ketamine binding site, ketamine physically blocks the NMDA channel (in much the same way as magnesium) and prevents glutamate from opening the channel . In other words, ketamine appears to treat depression by blocking calcium entry. (This blockade is also the basis for ketamine’s anesthetic property.) However, another perspective in the last section of this post presents an intriguing alternative explanation of ketamine’s antidepressant effect.
How does glutamate excitotoxicity bring about depression?
The Glutamatergic Hypothesis assumes that depression is ultimately caused by a breakdown in the functioning of glutamate communication in the brain. The various symptoms of depression are thought to arise mainly from the inability of different parts of the brain to interact normally. This section attempts to explain how these changes come about, drawing on research from non-human animals as well as humans.
Psychiatrists agree that Major Depressive Disorder is precipitated and maintained by psychological stress with depression-prone individuals having a heightened stress response. This stress response involves the increased release of adrenal glucocorticoids from the adrenal cortex (Moica et al, 2016). These hormones (such as cortisol in humans and corticosterone in some other species) are essential to life, affecting virtually every cell in the body. However, under conditions of stress, glucocorticoid release is substantially elevated. One effect of heightened release is to increase glucose availability, the preferred fuel of all cells of the body (hence the name, glucocorticoid). A second effect is to suppress inflammation in the body by dampening the immune system. In this way, glucocorticoids help individuals cope with the stress.
While adaptive in the short-term, an extended stress response is maladaptive. Continued high levels of glucocorticoids deplete the body’s energy resources and eventually impair, rather than help, the body’s functioning. The weakened immune system increases susceptibility to infection.
Glucocorticoids also readily cross the blood brain barrier where they affect both neurons and glia. One of the outcomes is that glutamate neurons increase their synaptic activity. Similar to events outside the brain, the increase in glutamate activity is likely beneficial in the short term, but in the longer term can lead to glutamate excitoxicity and depression.
Glutamate hyperactivity is also associated with a reduction in brain-derived neurotrophic factor (BDNF) in the brain’s extracellular fluid. BDNF is a protein synthesized by glia and neurons (Khoodoruth et al., 2022) and is released into the extracellular fluid, where it binds non-synaptic receptors on glutamate neurons. This binding promotes the health, neuroplasticity, and general functioning of glutamate neurons. Lowered extracellular BDNF further contributes to glutamate neuron dysfunction. In the hippocampus, BDNF also appears essential for neurogenesis (more about that below). In animals, the type of glutamate dysfunction seen in human depression can be corrected with BDNF administration directly into the brain, suggesting BDNF would be a powerful antidepressant! Unfortunately, BDNF does not cross the blood/brain barrier, preventing its practical use in humans.
However the causal relationship between glutamate activity and BDNF activity is complex (Gullyaeva, 2016). Animal research has shown that cause and effect can operate in both directions. That is, glutamate malfunctioning can cause BDNF levels to drop, while decreased BDNF levels can cause glutamate malfunctioning. So which occurs first in the chronology of depression is not clear. (This ambiguity as well as the clear relationship between BDNF and depression has given rise to a separate BDNF hypothesis of depression.) It is worth mentioning that BDNF decreases are also seen in other neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, multiple sclerosis and Huntington’s disease.
While there is some loss of the brain’s glutamate neurons during depression, the more significant effect appears to be a loss in glutamate synapses (as measured by a loss of dendritic spines). This loss results in a maladaptive remodeling (i.e. “rewiring”) of the glutamatergic circuitry both within and between various brain structures (Sancora et al., 2012). Again, the more critical factor is thought to be the impaired ability of the cortex, limbic system, and basal ganglia to work together in a coordinated fashion to regulate emotion and cognition.
Malfunctioning in glutamate neurons can cause dysfunction in other neurons as well. This dysregulation can, in turn, feed back on glutamate circuitry further contributing to glutamate dysfunction. For example GABAergic dysfunction also is seen during depression (Fogaca, 2023) suggesting that its normal inhibitory regulation of glutamate neurons is affected. Maladaptive glutamate functioning could also contribute to the low monoamine (serotonin, norepinephrine & dopamine) activity seen in depressed individuals, which in turn contributes to glutamatergic dysfunction. It seems likely that many other neurotransmitter systems also would be affected.
As mentioned earlier, glia cells are also adversely affected during depression. Since oligodendrocytes provide the myelin (“white matter”) that speeds up axonal action potentials, loss of myelin affects the speed and timing of glutamatergic communication. Also the nurturance of glutamate axons by myelin is disturbed.
Astrocyte dysfunction during depression contributes through its roles in glutamate removal from synapses, in preventing glutamate”leakage” out of the synapse, and also in the biosynthesis of both glutamate and GABA. Astrocytes also contain a substantial amount of glutamate extracted from glutamate synapses. Normally, this glutamate is converted to glutamine and transferred to neurons to participate in glutamate (and GABA) biosynthesis. (see Figure 1 for glutamate synthesis). However, during depression, some of this glutamate can “leak out” of dysfunctional astrocytes further contributing to glutamate’s excitotoxicity. Microglia also become increasingly activated during depression to help the brain cope with the increased levels of inflammation (Harcon et al., 2017).
Just as neurons communicate with each other, astrocytes, oligodendroglia and microglia also communicate with each other to coordinate their contributions to glutamate functioning. During depression this inter-glial communication can also become disturbed, further contributing to glutamate dysfunction (Harcon et al., 2017).
Some MRI or CAT scan studies report that depressed individuals experience a small shrinkage in certain brain areas, reflecting a loss of both grey and white matter. Although there are differences between studies, the affected areas can include the frontal lobe, hippocampus, temporal lobe, thalamus, striatum, and amygdala (Zhang et al., 2018). Zhang et al. (2018) suggests that the differences in different studies may be related to the length and severity of the subjects’ depression. Again, the shrinkage doesn’t appear to be caused as much by neuron loss as it is by the loss of glutamate terminals and dendrites (contributing to grey matter loss) and the loss of myelin (contributing to white matter loss).
Perhaps the most consistent finding is the shrinkage of the hippocampus (Malykhin & Coupland, 2015). Shrinkage by as much as 20% has been seen in some severely depressed patients (giving rise to the Hippocampal Hypothesis of Depression). However, the hippocampus is thought to have a unique feature that contributes to its shrinkage. Many neuroscientists believe the hippocampus to be the only area of the adult human brain that engages in neurogenesis (the birth of new neurons). All the other brain areas contain only neurons that were present at birth.
Neurogenesis leads to 2 neuronal populations in the hippocampus. One population resembles the long-lived neurons in other parts of the brain. If these neurons die, they are not replaced. The other population is shorter lived and is constantly being replaced by neurogenesis, keeping their numbers relatively constant throughout life. (It is thought that this population plays an important role in making the new synaptic connections throughout life to support the hippocampus’s role in memory formation.) During depression, the drop in BDNF levels causes hippocampal neurogenesis to become impaired. The resulting shrinkage is thought to be caused mainly by the short-lived neurons not being replaced as quickly as they are lost. The resulting dysfunction is also thought to give rise to the Alzheimers-like memory problems seen in many severely depressed individuals.
The good news is that brain shrinkage appears to be at least partially reversible by successful depression treatment. The bad news is that the more severe and long lasting the depression, the more refractory it is to available treatments.
Is Depression actually multiple disorders with overlapping symptoms?
One puzzling feature of unipolar depression is that patients often vary somewhat in primary and secondary symptoms. According to the DSM5 (the Diagnostic and Statistical Manual of the American Psychiatric Association), a patient must possess 5 or more of 8 depression symptoms to be diagnosed as suffering from Major Depressive Disorder (MDD). The patient must exhibit one of the 2 primary symptoms, either a depressed mood or anhedonia (i.e. an inability to experience reward). The remaining secondary symptoms can include weight loss or gain, feelings of worthlessness, feelings of guilt, fatigue, lack of concentration, anxiety, memory problems, and suicidal thoughts. The symptom variability seen from patient to patient has led to the idea that depression may not be a single disorder but rather multiple disorders with overlapping symptoms.
However the glutamatergic hypothesis can potentially explain different symptom profiles. The different symptoms, are to some extent, caused by malfunctioning in different parts of the brain, all of which depend upon glutamate functioning. It is possible that different brain areas have different susceptibilities to dysfunction in different patients, accounting for different profiles. At the same time, the idea of multiple disorders masquerading as a single disorder cannot be ruled out.
Implications of the Glutamatergic Hypothesis for treatment.
According to the glutamatergic hypothesis, the brain’s glutamate system is considered the final common pathway for all depression treatments (Sancora et al., 2012). Conventional thinking for the 3 major classes of antidepressants has been that ketamine affects glutamatergic dysfunction directly by blocking NMDA receptors. SSRI’s (e.g. fluoxetine, citaloram, escitalopram, and others) work by blocking serotonin reuptake transporters, and monoamine psychedelics (e.g. psilocybin, LSD and ayahuasca) work by activating a type of serotonin receptor (5-HT2A ).
But that may not be all, folks! Sanacora et al. (2012) cites evidence that drugs that interact with AMPA receptors, as well as drugs that interact with certain glutamate metabotropic receptors, may also have antidepressant qualities. In addition, Negrete-Diaz et al., (2022) suggest that kainate receptors might be involved in depression as well. Ideally, a drug, or drug combination, that addresses multiple aspects of glutamatergic functioning might work the best. If the technical issues of delivery could be solved, perhaps BDNF, which is beneficial for both neurons and glia, or a BDNF-analog drug would be ideal.
Just as I was getting ready to push the “publish” button for this post, I came across a recent publication by Moliner et al. (2023) that provides a potentially “paradigm-shifting,” alternative explanation of how antidepressants affect glutamateric circuitry! These authors provide evidence that the three major classes of antidepressant drugs all work by binding a type of BDNF receptor called TrkB (Tropomyosin receptor kinase B). In other words, the current antidepressant drugs may already be BDNF analogs of sorts! From this perspective, NMDA receptors, serotonin reuptake transporters, and 5-HT2A receptors would more likely be associated with antidepressant-drug side effects.
Consistent with this perspective, previous work (Casarotto et al., 2021) demonstrated that both SSRI’s and ketamine exhibit low affinity TrkB binding. However, Moliner et al. (2023) found that psilocybin and LSD, both of which have antidepressant properties, bind the TrkB receptor with a 1000-fold higher affinity (Moliner et al., 2023). Although direct comparisons of the effectiveness of different classes of antidepressant drugs are lacking, these findings seem congruent with subjective impressions of antidepressant effectiveness. For example, ketamine appears better than SSRI’s both in terms of speed of action (hours vs weeks) and in the number of patients that respond (many patients respond to ketamine that don’t respond to SSRIs). The monoamine psychedelics also act quickly in many patients unresponsive to SSRIs. However, while the ketamine antidepressant effects last around a week in most patients, the effects of the monoamine psychedelics often appear to last for months or longer.
However, Moliner et al., 2023, found that psilocybin and LSD don’t work exactly like BDNF. Rather these drugs bind the TrkB receptor at a different site than BDNF allowing them to serve as allosteric modulators of the TrkB receptor. In so doing, these drugs make the TrkB receptor more sensitive to endogenous BDNF by slightly changing the shape of the TrkB receptor (Moliner et al., 2023). Presumably this increased sensitivity would allow the low levels of BDNF present during depression to be sufficient to correct the glutamatergic dysfunction and treat the depression. Furthermore you would expect drugs that bind TrkB with high affinity (such as psilocybin) to produce quicker and more lasting relief than drugs that bind with low affinity (such as SSRIs). Furthermore, if the antidepressant and psychedelic binding sites of the psychedelic antidepressant drugs are distinct, it might be possible to develop powerful new antidepressant drugs that are not psychedelic!
However the significant positive correlations between the antidepressant effects of psilocybin, ayahuasca, and ketamine and their psychedelic effects (Mathai et al.,2020; Ko et al., 2022) could be used to argue against the likelihood of developing non-psychedelic antidepressants. These correlations don’t invalidate the therapeutic importance of TrkB binding. However, if the correlations are caused by the same part of the molecule binding both the “psychedelic” receptor site and the TrkB receptor, it may not be possible to separate the two effects.
Further complicating the situation, another recent finding (Sanacora, & Colloca, 2023) is consistent with ketamine’s psychedelic effect causing its antidepressant activity. In this study, depressed patients were ingeniously prevented from experiencing ketamine’s psychedelic effect by administering it when the patients were unconscious from general anesthesia (for surgery unrelated to their depression). Under these circumstances, ketamine was no better than a placebo at reducing depression symptoms. How this finding can be reconciled with the hypothesized importance of TrkB binding isn’t clear.
It would be very informative to similarly “mask” the psychedelic effect of psilocybin during depression treatment. Unfortunately such an attempt would be difficult (and probably unethical) since the duration of psilocybin’s psychedelic effect (around 6 hours) is much longer than that of ketamine (around an hour).
It’s now time to throw up our collective hands and utter the common response to ambiguous scientific situations, “more research is needed!”
Conclusion.
The evidence is compelling that depression is related to the malfunctioning of the brain’s glutamateric circuitry. At the same time questions remain as to exactly how antidepressant drugs interact with this circuitry. It will be interesting to see how this all plays out over the next few years. Stay tuned!
References Consulted.
All the references below contain information that made contributions to my post.
Alnefeesi, Y., Chen-Li, D., Krane, E., Jawad, M. Y., Rodrigues, N. B., Ceban, F., . . . Rosenblat, J. D. (2022). Real-world effectiveness of ketamine in treatment-resistant depression: A systematic review & meta-analysis. Journal of Psychiatric Research, 151, 693-709. doi:10.1016/j.jpsychires.2022.04.037
Casarotto, P. C., Girych, M., Fred, S. M., Kovaleva, V., Moliner, R., Enkavi, G., . . . Castren, E. (2021). Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell, 184(5), 1299-1313.e19. doi:10.1016/j.cell.2021.01.034
Fogaca, M. V., & Duman, R. S. (2019). Cortical GABAergic dysfunction in stress and depression: New insights for therapeutic interventions. Frontiers in Cellular Neuroscience, 13, 87. doi:10.3389/fncel.2019.00087
Gulyaeva, N. V. (2017). Interplay between brain BDNF and glutamatergic systems: A brief state of the evidence and association with the pathogenesis of depression. Biochemistry.Biokhimiia, 82(3), 301-307. doi:10.1134/S0006297917030087
Haroon, E., Miller, A. H., & Sanacora, G. (2017). Inflammation, glutamate, and glia: A trio of trouble in mood disorders. Neuropsychopharmacology : Official Publication of the American College of Neuropsychopharmacology, 42(1), 193-215. doi:10.1038/npp.2016.199
Hawkins, R. A. (2009). The blood-brain barrier and glutamate. The American Journal of Clinical Nutrition, 90(3), 867S-874S. doi:10.3945/ajcn.2009.27462BB
Khoodoruth, M. A. S., Estudillo-Guerra, M. A., Pacheco-Barrios, K., Nyundo, A., Chapa-Koloffon, G., & Ouanes, S. (2022). Glutamatergic system in depression and its role in neuromodulatory techniques optimization. Frontiers in Psychiatry, 13, 886918. doi:10.3389/fpsyt.2022.886918
Ko, K., Knight, G., Rucker, J. J., & Cleare, A. J. (2022). Psychedelics, mystical experience, and therapeutic efficacy: A systematic review. Frontiers in Psychiatry, 13, 917199. doi:10.3389/fpsyt.2022.917199
Malykhin, N. V., & Coupland, N. J. (2015). Hippocampal neuroplasticity in major depressive disorder. Neuroscience, 309, 200-213. doi:10.1016/j.neuroscience.2015.04.047
Mathai, D. S., Meyer, M. J., Storch, E. A., & Kosten, T. R. (2020). The relationship between subjective effects induced by a single dose of ketamine and treatment response in patients with major depressive disorder: A systematic review.Journal of Affective Disorders, 264, 123-129. doi:10.1016/j.jad.2019.12.023
Moica, T., Gligor, A., & Moica, S. (2016). The relationship between cortisol and the hippocampal volume in depressed patients – a MRI study. Procedia Technology, 22, 1106-1112.
Moliner, R., Girych, M., Brunello, C. A., Kovaleva, V., Biojone, C., Enkavi, G., . . . Castren, E. (2023). Psychedelics promote plasticity by directly binding to BDNF receptor TrkB. Nature Neuroscience, 26(6), 1032-1041. doi:10.1038/s41593-023-01316-5
Moriguchi, S., Takamiya, A., Noda, Y., Horita, N., Wada, M., Tsugawa, S., . . . Nakajima, S. (2019). Glutamatergic neurometabolite levels in major depressive disorder: A systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Molecular Psychiatry, 24(7), 952-964. doi:10.1038/s41380-018-0252-9
Onaolapo, A. Y., & Onaolapo, O. J. (2021). Glutamate and depression: Reflecting a deepening knowledge of the gut and brain effects of a ubiquitous molecule. World Journal of Psychiatry, 11(7), 297-315. doi:10.5498/wjp.v11.i7.297
Pitsillou, E., Bresnehan, S. M., Kagarakis, E. A., Wijoyo, S. J., Liang, J., Hung, A., & Karagiannis, T. C. (2020). The cellular and molecular basis of major depressive disorder: Towards a unified model for understanding clinical depression. Molecular Biology Reports, 47(1), 753-770. doi:10.1007/s11033-019-05129-3
Sanacora, G., & Colloca, L. (2023a). Placebo’s role in the rapid antidepressant effect. Nature Mental Health, 1(11), 820-821. doi:10.1038/s44220-023-00141-w
Sanacora, G., Treccani, G., & Popoli, M. (2012). Towards a glutamate hypothesis of depression: An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology, 62(1), 63-77. doi:10.1016/j.neuropharm.2011.07.036
Sapolsky, R. M. (2001). Depression, antidepressants, and the shrinking hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 98(22), 12320-12322. doi:10.1073/pnas.231475998
Sheline, Y. I. (2011). Depression and the hippocampus: Cause or effect? . Biol. Psychiatry, 70(4), 308-309.
van Velzen, L. S., Kelly, S., Isaev, D., Aleman, A., Aftanas, L. I., Bauer, J., . . . Schmaal, L. (2020). White matter disturbances in major depressive disorder: A coordinated analysis across 20 international cohorts in the ENIGMA MDD working group. Molecular Psychiatry, 25(7), 1511-1525. doi:10.1038/s41380-019-0477-2
Wang, H., He, Y., Sun, Z., Ren, S., Liu, M., Wang, G., & Yang, J. (2022). Microglia in depression: An overview of microglia in the pathogenesis and treatment of depression. Journal of Neuroinflammation, 19(1), 132-0. doi:10.1186/s12974-022-02492-0
Zhang, F., Peng, W., Sweeney, J. A., Jia, Z., & Gong, Q. (2018). Brain structure alterations in depression: Psychoradiological evidence. CNS Neuroscience & Therapeutics, 24(11), 994-1003. doi:10.1111/cns.12835





























{kind=link}