Introduction.
Sleepiness is a powerful force compelling us to go to sleep each night. While sleepiness can be resisted for extended periods, it always wins out in the end. The negative consequences are just too negative. In fact, in animal research, extreme sleep deprivation has sometimes proven fatal.
According to the Guinness Book of World Records, the official human record for staying awake of 11 days and 25 minutes was set in 1964 by a then 17-year-old Randy Gardner. In addition to becoming incredibly sleepy, Gardner experienced sensory, emotional, and cognitive deficits as his sleep deprivation progressed. Fortunately, he suffered no lasting effects. However, after granting Gardner the world record, the Guinness Book of World Records decided, for ethical (and probably legal) reasons, to no longer consider world records for staying awake. While Gardner’s record has since been “unofficially” broken, such attempts are discouraged by sleep experts!
This post will examine the neural processes that cause us to become sleepy. However, these processes all work by inhibiting wakefulness, the default state. So, in order to understand the causes of sleepiness, you must first understand the causes of wakefulness.
Brainstem Mechanisms Promoting Wakefulness.
Both sleepiness and wakefulness arise from the functioning of the cerebral cortex. At the same time, the cortex’s management of these states of consciousness is guided by the brainstem. The most important brainstem structures are located in the reticular system, a diffuse, polysynaptic pathway running up the center of the brain. At one time the reticular system was thought to be an alternative sensory system because it receives input from all the different senses, including vision, taste, smell, touch, temperature, etc. However we now know its “job” is not to process the nature of this sensory input, but rather to act as an “early warning system” to keep the cortex awake and alert when important sensory information needs to be acted upon.
Three different reticular activating system structures contribute to wakefulness including the Medial Pontine Reticular Formation, the Locus Coeruleus, and the anterior Raphé Nuclei. All become active under similar circumstances and produce different, but complementary, effects. A fourth area, just anterior to the Reticular Activating system, the Tuberomammillary Nucleus of the hypothalamus, also contributes.
All 4 structures affect wakefulness and arousal through the release of specific neurotransmitters into the cortex. For all these neurotransmitters, release is highest when awake and behaviorally active, lower when awake and nonactive, even lower during NREM sleep, and except for acetylcholine, lowest during REM sleep. Thus the effects of these neurotransmitters are greatest when the need for wakefulness is highest.
In my teaching I found it was often easier for students to understand brainstem neuroanatomy when displayed on a simpler primitive vertebrate brain than on a human brain (with its twists and turns). So that is what I do here. A more detailed description of primitive vertebrate neuroanatomy and its relationship to human neuroanatomy can be found in another post.
Medial Pontine Reticular Formation.
The neurons of the Medial Pontine Reticular Formation originate in the middle of the pons and use acetylcholine as their neurotransmitter. However, rather than projecting directly to the cortex, their input is relayed by 2 intervening structures. In one pathway the Nucleus Basalis serves as a relay, and in the other pathway, 2 thalamic nuclei (the medial and intralaminar nuclei) serve as a relays (See Figure 2). However, the “relay” neurons also release acetylcholine as their neurotransmitter. Presumably if one pathway fails, the other can take over and insure that this important process is carried out.

Acetylcholine exerts its arousing effects by inhibiting a type of cortical neuron known as a “pacemaker” neuron. While pacemaker neurons represent only a small fraction of all cortical neurons, they have the unusual ability to synchronize the action potentials of the many hundreds of other neurons in their vicinity. The pacemaker neurons are most influential during the deepest stages of NREM sleep, when acetylcholine input is very low. At this time, the pacemaker neurons cause the cortex to exhibit the high-amplitude, low-frequency EEG characteristic of sleep. This role was described in detail in a previous post.
Upon awakening in the morning, acetylcholine release begins inhibiting the pacemaker neurons which, in turn, causes the cortical EEG to become more desynchronized, exhibiting a lower amplitude and higher frequency. Throughout the day, whenever the person encounters a challenging situation, acetylcholine release further enhances cortical desynchronization. Even mild challenges such as deciding whether to have pancakes or oatmeal for breakfast cause some increase. (That’s actually a difficult challenge for me!) The more challenging the “problem,” the more activated the reticular formation, and the more desynchronized the cortex.
When awake, cortical desynchronization enhances the cortex’s ability to engage in cognitive processing, allowing the person to better solve the day’s problems. (The reason why cortical desynchronization enhances cognitive processing is explained in an earlier post). Conversely, should the effects of acetylcholine be prevented, cognitive ability would be diminished. Two drugs that do just that are scopalamine and atropine. Both are acetylcholine-receptor blockers and can produce cognitive and memory deficits that mimic Alzheimer’s Disease. Fortunately the drug effects are only temporary! On the other hand, the permanent symptoms of Alzheimer’s Disease are caused by the death of acetylcholine-releasing neurons in the Nucleus Basalis (as well acetylcholine neurons in the lateral septum and diagonal band of Broca that target the hippocampus). As Alzheimer’s disease progresses, other types of cortical neurons die as well.
In summary, acetylcholine release into the waking cortex causes the cortical desynchrony that both promotes wakefulness and allows us to use our cognitive abilities to their fullest.
Anterior Locus Coeruleus.
A second reticular activating system structure promoting a different aspect of wakefulness is the Anterior Locus Coeruleus of the dorsal pons (See Figure 3). The Anterior Locus Coeruleus does so by releasing norepinephrine into the cortex via a fiber tract called the dorsal noradrenergic bundle (also called the dorsal tegmental bundle). (Other parts of the Locus Coeruleus innervate other areas of the brain and spinal cord.)
Norepinephrine is the neurotransmitter of the sympathetic branch of the autonomic nervous system whose role is to prepare vertebrates to deal with emotion-provoking situations (which in the extreme are referred to as “fight or flight” situations). The degree of release is proportional to the intensity of emotion experienced.

Norepinephrine release into the cortex causes a state of vigilance (i.e. extreme attentiveness to sensory input). Vigilance is very adaptive in emotion-provoking situations because it makes you very alert and causes you to pay careful attention to everything going on around you. Norepinephrine elevations also diminish other motivations, such as hunger and pain, that might divert you from the important issues at hand.
Although cocaine and amphetamine are used recreationally mainly for their rewarding effects (caused by dopamine elevations), their stimulant effects depend upon norepinephrine elevations. For example, amphetamine’s ability to elevate brain norepinephrine underlies its (mostly illegal) uses as an appetite suppressant (diet pills), as an all-nighter study aid for college students, and as a driving aid for long-haul truck drivers. My father was a P38 fighter pilot in World War II and he told me that he used to take some little pills before his missions. He didn’t know what they were, but it’s pretty easy to guess!
During sympathetic arousal, many body organs outside the brain also receive enhanced norepinephrine neurotransmitter input from sympathetic motor neurons to further insure adaptive responses to emotional situations. These elevations increase muscle tone, heart rate, and blood pressure, while also optimizing energy release from both fat and carbohydrate stores. These peripheral neurotransmitter effects are further enhanced by the adrenal medulla, which simultaneously releases norepinephrine (and epinephrine) into the blood stream adding to the effects of sympathetic neuron release. Consequently, outside the brain, norepinephrine is both the neurotransmitter, and the hormone, of sympathetic arousal.
Anterior Raphe Nuclei.
The Raphé Nuclei are a series of 8 reticular system nuclei that are the primary source of the brain neurotransmitter, serotonin. The more anterior nuclei service the cortex while the remaining service the rest of the brain and the spinal cord (See Figure 4). Virtually all areas of the brain and spinal cord receive input. Like the two previous arousal centers, the Raphé nuclei are most active when awake and physically active.
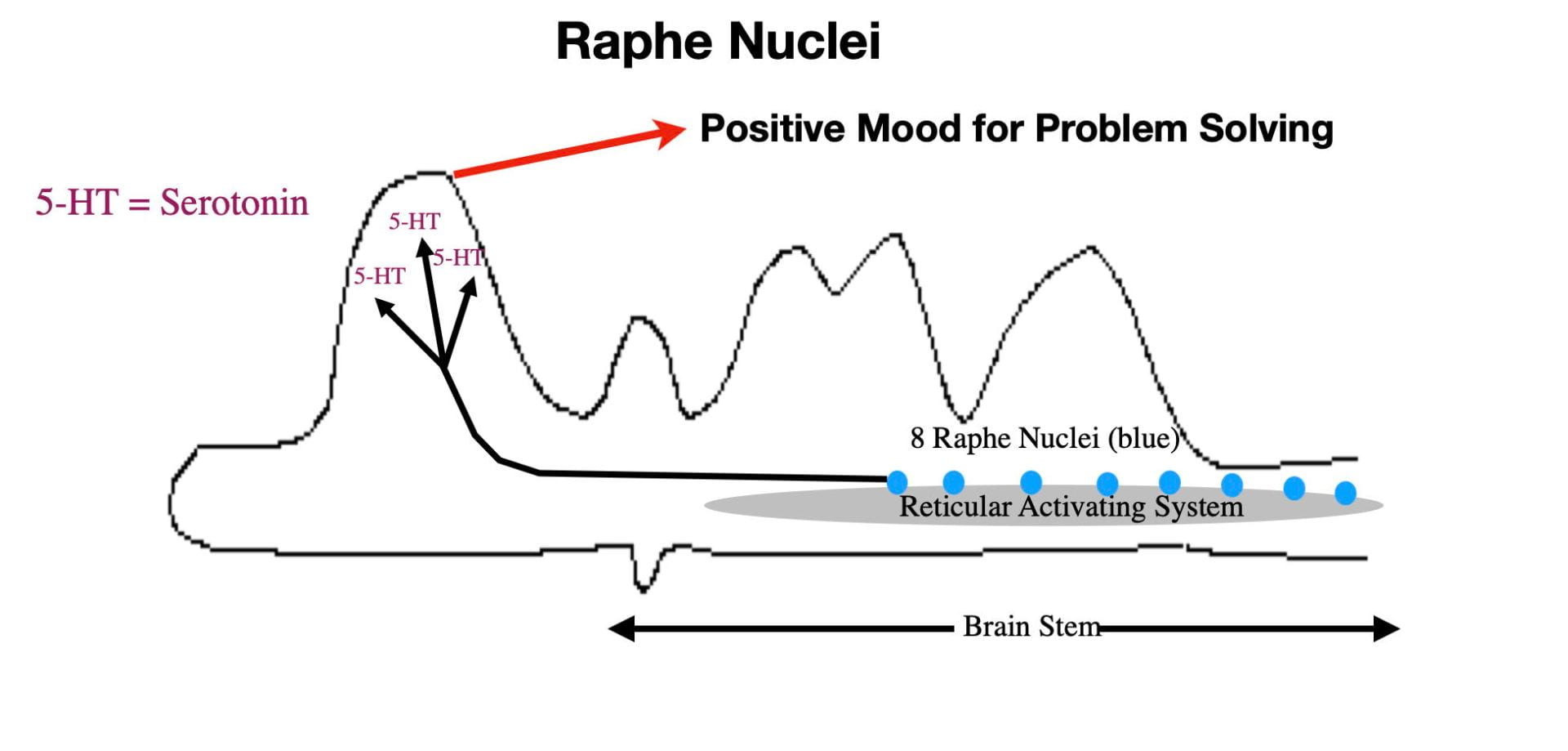
The Raphé Nuclei make several contributions to our consciousness through serotonin elevations in the cortex. For normal individuals, naturally occurring serotonin elevations help provide the mental energy and mindset that promotes adaptive responses to the challenges of the day. In contrast, abnormally low levels of serotonin are associated with depression. In addition to feeling sad, depressed individuals often suffer from high levels of anxiety, low energy levels, slowness of movement, chronic fatigue, and a sense of hopelessness. As a result depressed individuals often don’t respond appropriately to the day’s challenges. By boosting brain serotonin levels, the first-line antidepressant drugs (selective serotonin reuptake inhibitors (SSRIs)) restore more normal behavior in depressed individuals. Although far from perfect, SSRI’s do allow more normal functioning in many (but not all) depressed patients.
However, another contribution may also be important. Just before muscle movements occur, serotonin neurons show micro bursts of serotonin release throughout the brain and spinal cord. This led to the idea that one of their roles may be to help the central nervous system to carry out movement. If true, this would also enhance our ability to respond behaviorally to the normal challenges of the day. Reduced serotonin secretion in depressed individuals may also contribute to their movement deficiencies.
Tuberomammillary Nucleus.
Neurons of the Tuberomammillary Nucleus, located just anterior to the reticular activating system, in the posterior portion of the hypothalamus, are the primary source of the neurotransmitter, histamine. A fiber tract from the Tuberomammillary Nucleus connects directly with the cortex where histamine release produces wakefulness and arousal in a fashion similar to acetylcholine (see Figure 5). As with the other neurotransmitters of arousal, histamine is released mainly when the brain needs to be alert and cognitively active.
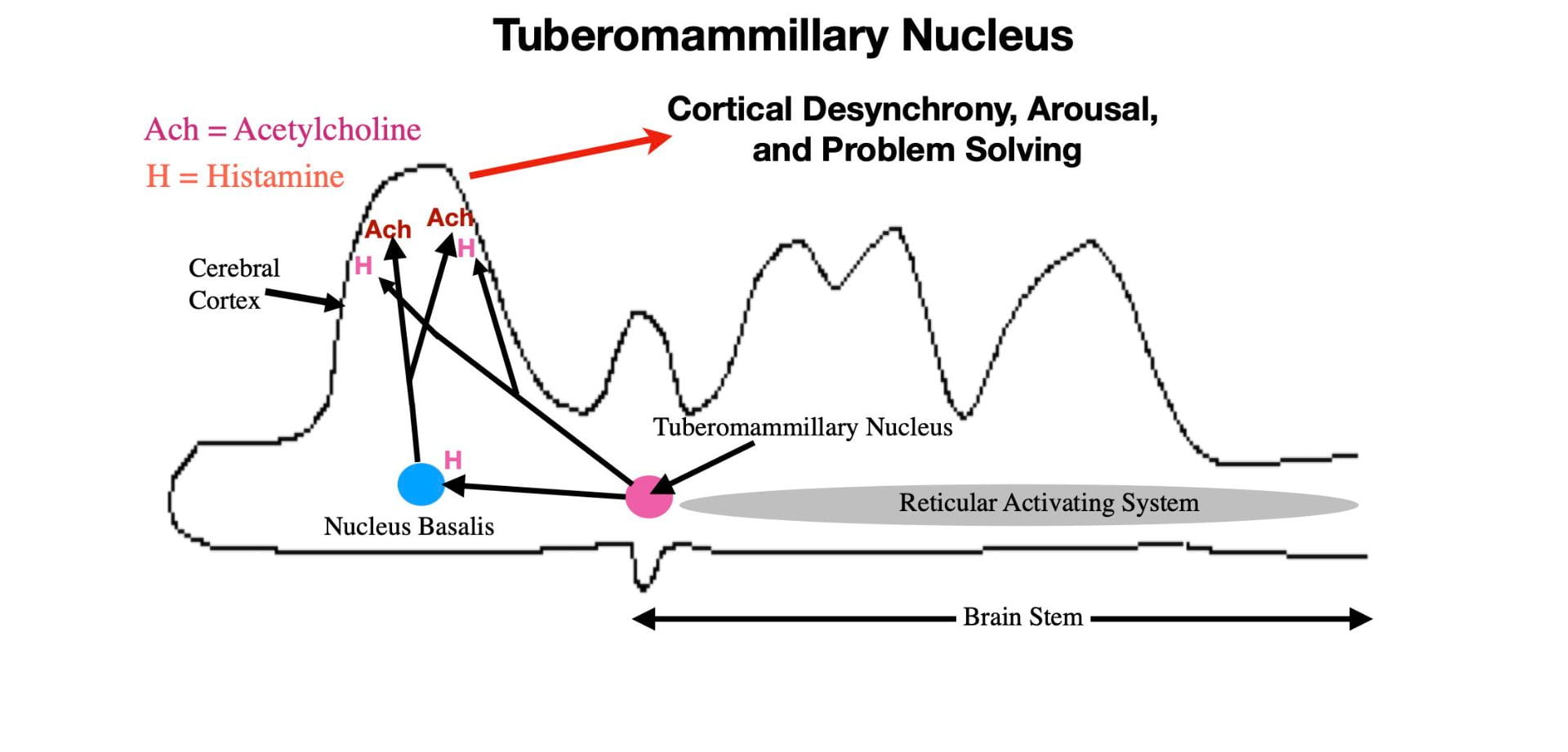
As expected, blocking the effects of cortical histamine produces sleepiness. Antihistamines are drugs taken by cold and allergy sufferers to reduce nasal congestion. These drugs provide relief by blocking histamine receptors in the nasal cavity. The newer generation antihistamines can not cross the blood brain barrier and thus do not affect the histamine receptors in the brain. However the older generation antihistamines do cross the blood brain barrier and, as predicted, their principal side effect is drowsiness. In fact, these older generation antihistamines are now recommended for use only at night and are also sold as over-the-counter sleeping aids.
The Tuberomammillary Nucleus also releases histamine into the Nucleus Basalis causing the Nucleus Basalis to release acetylcholine into the cortex. Thus in addition to arousing the cortex directly through histamine release, the Tuberomammillary Nucleus also arouses the cortex indirectly through promoting acetylcholine release (See figure 5).
In summary, all 4 brainstem inputs into the cortex promote alertness and enhance cognitive functioning in a complementary fashion and optimize adaptive responses to important sensory input.
Neural Mechanisms Promoting Sleep Onset.
The previously described neurotransmitter inputs into the cortex cause wakefulness and arousal to be the default cortical state. Sleepiness arises from the inhibition of these arousal mechanisms. Three processes known to inhibit the brainstem arousal systems are 1) the circadian buildup of adenosine in the extracellular fluid of the brain, 2) Increased activity in the Nucleus of the Solitary Tract, and 3) Increased activity in the Median Preoptic Nucleus.
Adenosine Buildup In the Extracellular Fluid.
Perhaps the most important factor normally causing the evening’s sleepiness is the buildup, over the course of the day, of a metabolic waste product called adenosine. When you wake up in the morning, brain adenosine levels are low and then gradually increase over the course of the day. This buildup promotes sleepiness. However, once asleep, the brain “disposes” of this waste product and the cycle repeats itself the next day.
In addition to being a waste product, adenosine is also an inhibitory neurotransmitter utilized by a small fraction of the brain’s neurons. (Reminds me of a funny SNL skit many years ago where a salesman was selling a product that was both a mouthwash and a floor polish). So it is not surprising that some of the brain’s neurons have adenosine receptors. However adenosine receptors can be bound not only by adenosine, the neurotransmitter, but also by adenosine, the waste product. It just so happens that the acetylcholine neurons of the Thalamus and Nucleus Basalis are rich in adenosine receptors. Over the day, extracellular adenosine increasingly binds these receptors, inhibiting acetylcholine release into the cortex, and by the end of the day, promoting cortical synchrony and sleepiness.

So…where does the waste adenosine come from? Adenosine is a metabolic breakdown product of two common and very important molecules. The first is adenosine triphosphate (ATP), which the mitochondria use as the energy source to drive intracellular metabolism. The other source is cyclic adenosine monophosphate (cAMP), a very common intracellular messenger. Both of these molecules are critically important to neuronal functioning and have the highest rate of use and breakdown during the day when metabolism is highest. As a consequence, the extracellular adenosine progressively builds up in the brain’s extracellular fluid over the course of the day, binding more and more adenosine receptors, and eventually causing sleepiness.
However, in addition to serving as a neurotransmitter and a waste product, adenosine also is an important precursor for synthesizing ATP and cAMP. At night, particularly during NREM sleep when brain metabolism drops, the brain is able to recycle, or dispose of, much of the extracellular adenosine, allowing the cycle to begin again the next day.
This explanation also helps to understand the effects of my favorite drug that I use every morning! Because caffeine is an adenosine receptor blocker, it keeps you awake and alert and able to write neuroscience posts. Caffeine also mildly elevates dopamine in the brain’s reward circuitry, adding to its desirable effects.
Nucleus of the Solitary Tract.
Another important sleep center is the Nucleus of the Solitary Tract (NST). (I’m not sure where neuroscientists get these strange names.) As seen in figure 6, the NST facilitates sleep onset by inhibiting the Medial Pontine Reticular Formation. This inhibition reduces acetylcholine input into the cortex, which desynchronizes the cortex, promoting sleep.

The Nucleus of the Solitary Tract is in turn heavily influenced by input from an unusual cranial nerve, the Vagus Nerve (see figure 7). Like the other 11 cranial nerves, the Vagus Nerve has both motor and sensory fibers that service parts of the head. But unlike the other cranial nerves, the Vagus Nerve also has sensory and motor connections with many parts of the lower body. This sensory input allows the brain to monitor activities in the viscera (digestive system, heart, and lungs) and in the skin. This input can, in turn, influence sleepiness.
For example, Vagus Nerve input into the NST is thought to underly the sleepiness occurring after a big meal. In this case, a full stomach is detected by both stretch and nutrient receptors which convey this information to the brain via the Vagus Nerve. This input activates the Nucleus of the Solitary Tract to promote sleepiness. This sleepiness is adaptive because the resulting inactivity enhances digestion. In fact, taking a nap after a big meal is common in some cultures.
Vagus Nerve input to the Nucleus of the Solitary Tract also explains why parents rock babies to sleep or, as I sometimes used to do, take them for a car ride. In both cases, mechanoreceptors in the skin are stimulated by the gentle skin vibrations, which communicate this information to the brain via the vagus nerve, causing sleepiness. Babies sleep a lot more than adults because this allows them to put their energy into growth rather than into physical activity. While this “sleepiness reflex” is pronounced in babies, I’ve also known some adults who always fall asleep on long car rides.
Median Preoptic Nucleus.
The Median Preoptic Nucleus is part of the Preoptic Area just anterior to the hypothalamus, another brain area important in regulating sleep. (Some neuroanatomists classify the Preoptic Area as part of the hypothalamus.)

The initial evidence that the Median Preoptic Nucleus is a sleep center was that damage to this area causes insomnia. We now know that the Median Preoptic Nucleus induces sleep by inhibiting the various arousal centers decreasing the release of acetylcholine, norepinephrine, and serotonin into the cortex. The resulting neurotransmitter decrease promotes sleepiness.
The Median Preoptic Nucleus also plays an important role in mammalian thermoregulation suggesting that this structure may be important in causing the sleepiness that results from a rise in core body temperature. Falling asleep in my classroom in the late Spring, before the University turns on the air conditioners, is not very adaptive in terms of doing well on my exams. However, sleeping while having a fever is definitely adaptive by helping your body better use its energy to combat whatever is causing the fever.
Falling Asleep
Each night, as the various brainstem arousal mechanisms reduce the release of acetylcholine, norepinephrine, serotonin, and histamine into the cortex, you become more and more sleepy and eventually fall asleep. Paralleling the gradual reductions in neurotransmitter release and in changing EEG, falling asleep appears to be gradual as well.

Just before you fall asleep at night, during Stage W (see figure 8), your cortex is exhibiting the alpha waves characteristic of relaxed wakefulness. The first stage of sleep that you enter is NREM1 sleep. Here the alpha waves become increasingly interspersed with theta waves. These EEG oscillations cause you to drift back and forth between different levels of environmental awareness. In fact, if you “awaken” someone during NREM1 sleep, around 90% will tell you, incorrectly, that they weren’t really asleep.
The next stage of sleep is NREM2. Here theta waves have become the dominant EEG waveform. In addition, the occurrence of two additional wave forms, sleep spindles and K complexes, help define this stage of sleep. Sleep spindles are short bursts of beta waves and K complexes are short bursts of delta waves. A sleep researcher, upon seeing sleep spindles and K complexes, can be almost certain that the person is now asleep. Although more difficult to wake than in NREM1 sleep, around 40% of individuals awakened from NREM2 sleep will incorrectly tell you that they were awake and not really sleeping.
When you enter the deepest and most important NREM stage, NREM3 (also known as slow wave sleep), the brain waves have transitioned to mainly delta waves with some theta waves intermixed. Individuals are more difficult to wake up in NREM 3. When awoken, these individuals are often groggy and disoriented, and will almost always admit to being asleep.
The time from falling asleep to entering NREM3 sleep averages around 15 minutes. As measured by either EEG or by difficulty in awakening the sleeper, the journey from Stage W to NREM3 is gradual.
REM Sleep
After being asleep for an hour or so, the person passes from NREM sleep into the first REM sleep bout of the night. However, unlike the gradualness of passing through the various NREM stages, the transition from NREM to REM sleep is abrupt.
The shift back and forth between NREM sleep and REM sleep over the course of the night is thought to occur because of a “flip/flop” switch mechanism that operates almost instantaneously. The subject of the next post will be the brainstem neuroanatomy that causes this abrupt shift from NREM to REM sleep and vice versa.


{kind=link}