Introduction.
Some people are clearly more addiction prone. While both environmental and biological factors contribute, a recent publication by Li et al (2021) provides important new information about the role of brain biology. In prior research by other investigators, several brain areas had been implicated in protecting against addiction, as well as a role for the neurotransmitter, serotonin. However, the exact areas, and how serotonin interacts with these areas, were not known. Li et al’s research confirms the importance of serotonin, as well as identifies the dorsal striatum as the brain area where serotonin exerts its effects. The findings also have broader implications for understanding individual differences in addiction liability.
To provide context, some background material will be presented first.
Addiction is caused by highly maladaptive learning.
Learning is operationally defined as a relatively permanent change in behavior brought about by experience. By this definition, drug addiction is a type of learning. What is learned is the expectation of reward. With repeated drug use, this expectation intensifies, making drug use increasingly difficult to resist. When the urge becomes nearly irresistible, the individual is said to be addicted.
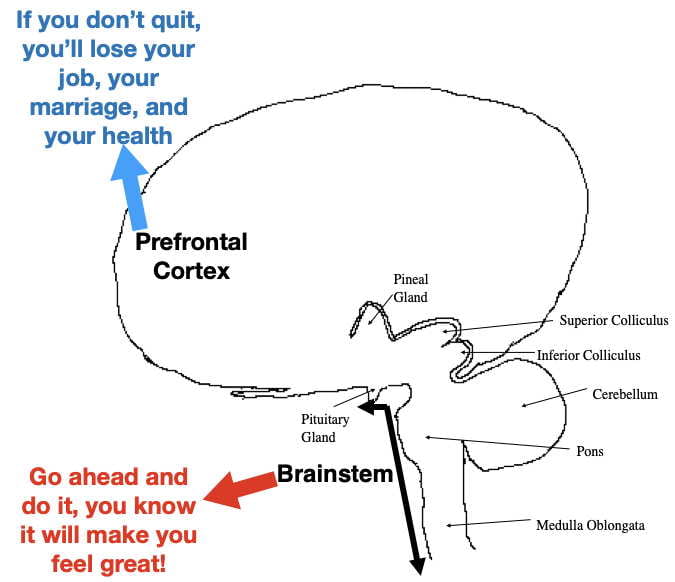
Unfortunately, once established, addiction appears to play by different rules than many other types of learning. For example, most learned behaviors are abandoned when they later become associated with bad outcomes. Not so for addiction. Many addicts continue drug use after their health is impaired, their marriages are destroyed, their jobs lost, or they find themselves in legal jeopardy. Addicts are often aware that their addiction is bad for them, they just can’t help themselves.
One possible explanation, explored in a previous post, is that the expectation of drug reward may be encoded, at least in part, in primitive, unconscious parts of the lower brainstem, resistant to influence by conscious thought processes (Fig 1). However, not totally resistant. While around 20% of human cocaine users eventually become addicted, the other 80% seem able to resist the urges underlying compulsive drug use.
The process of discontinuing a learned behavior is referred to by psychologists as extinction. In the parlance of psychology, the problem of an addicted drug user is an inability to extinguish drug use when it leads to negative outcomes. Extinction is considered by psychologists to be caused by new learning superimposed upon, and overriding, older learning. The older learning (expectation of drug reward) doesn’t go away, but rather the new learning (that taking the drug has bad consequences) prevents the old learning from being acted upon. Li et al’s research addresses why addicted users are unable to extinguish their drug use as circumstances become negative.
The reward pathway of the brain.

One of the great discoveries of neuroscience is that a final common brain pathway, highly similar in all vertebrates, plays a central role in all forms of reward. Natural reinforcers such as food, drink, sex, warmth, social interactions, etc. are all experienced as rewarding because they activate this pathway, as do rewarding drugs and electrical stimulation of certain parts of the brain.
The reward pathway originates in the ventral tegmental area, whose dopamine-releasing axons project to the nucleus accumbens (See Figure 2). Dopamine elevations in the nucleus accumbens causes the experience of reward. However, the manner in which drugs activate this pathway varies. Cocaine and amphetamine activate the nucleus acumen’s dopamine reward system directly, while other addictive drugs (such as opiates, alcohol, benzodiazepines and THC) initially act through other neurotransmitter systems that indirectly lead to dopamine elevations in the nucleus accumbens. Highly rewarding drugs, such as cocaine or heroin, generally cause more intense dopamine elevations than natural reinforcers which contribute to the drug’s addictiveness.
How Does Cocaine Activate the Reward Pathway?
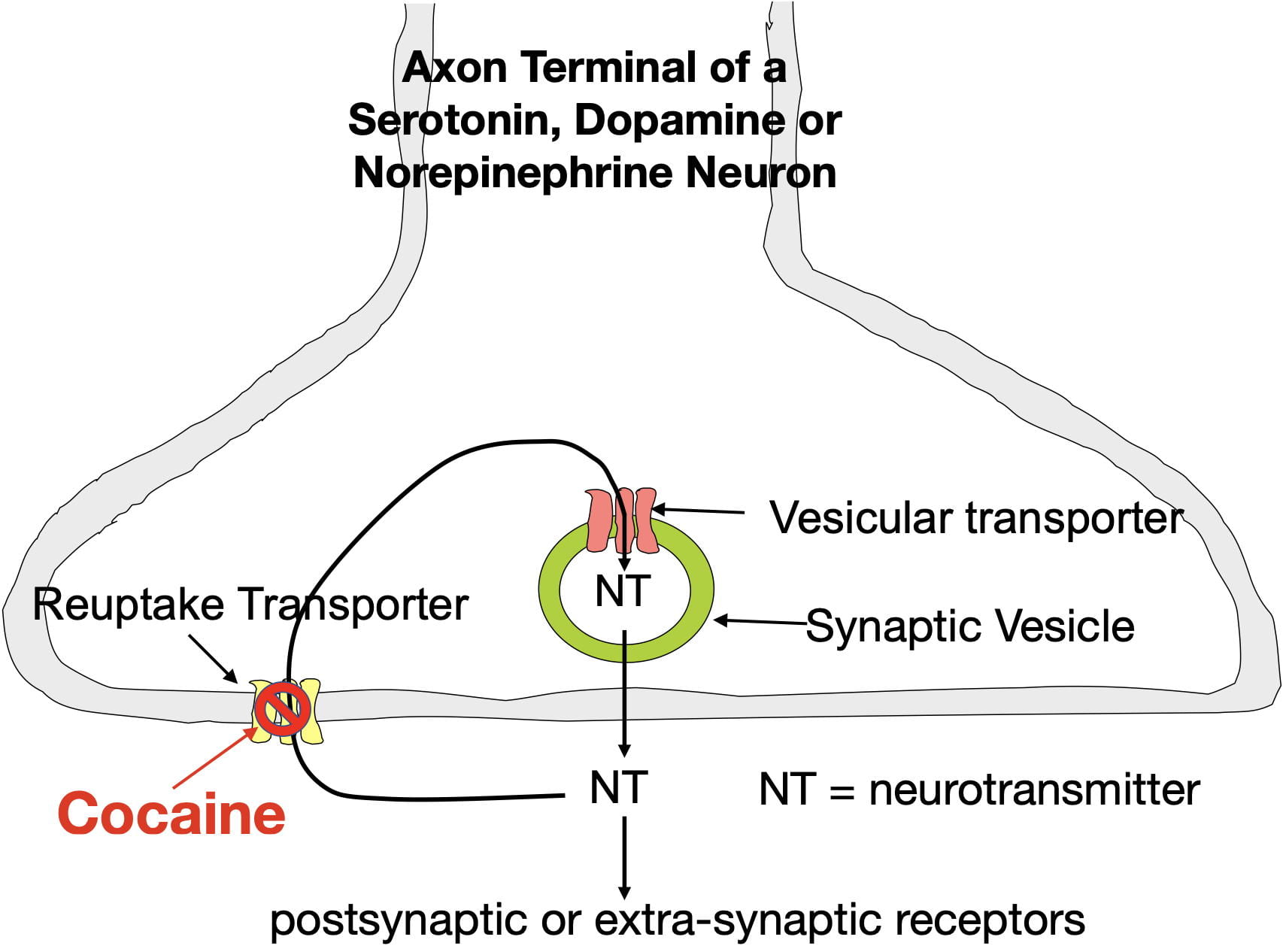
Cocaine produces the sensation of reward by blocking dopamine reuptake transporters in the nucleus accumbens. These membrane transporters serve to remove extracellular dopamine from the synapse by actively moving it back inside the neuron where it can be reused (see figure 3) . Cocaine disables this transporter. As a result, cocaine causes dopamine to build up in nucleus accumben’s synapses. The enhanced binding due to this buildup causes cocaine to be experienced as rewarding.
However, an important issue that I will return to later, is that cocaine (administered by injecting, snorting, smoking, or eating) goes everywhere in the brain where, in addition to causing reward, it produces other effects. For example, dopamine elevations in the dorsal striatum contribute to an increase in motor activity.
Cocaine also blocks norepinephrine and serotonin reuptake transporters throughout the brain causing the the synaptic buildup of these neurotransmitters as well. The resulting norepinephrine elevations cause sympathetic arousal (i.e. the fight or flight response) which contributes to heightened activity and also puts your body on high alert. On the other hand, the serotonin elevations provide an antidepressant effect (however, cocaine’s addictiveness and other effects make it undesirable as an useful antidepressant). Despite cocaine being highly addictive, Li et al found that cocaine-caused serotonin elevations paradoxically reduce cocaine’s addiction liability in mice (more about that later).
Recreational cocaine users take cocaine mainly for its rewarding properties. While the reward from repeated cocaine use is necessary for an addiction to develop, it is not sufficient. The research by Li et al provide evidence that other brain areas influence whether a cocaine “addiction” actually develops.
A Mouse Model of Cocaine Addiction.
Humans are not the only organisms capable of drug addiction. Nonhuman animals, such as mice and rats, like the same drugs as humans and, if given the opportunity, will self administer them. With continued administration, some eventually develop the compulsive, uncontrollable use characteristic of drug addicts. In some cases, these animals, like humans, have died from self-administered overdoses.
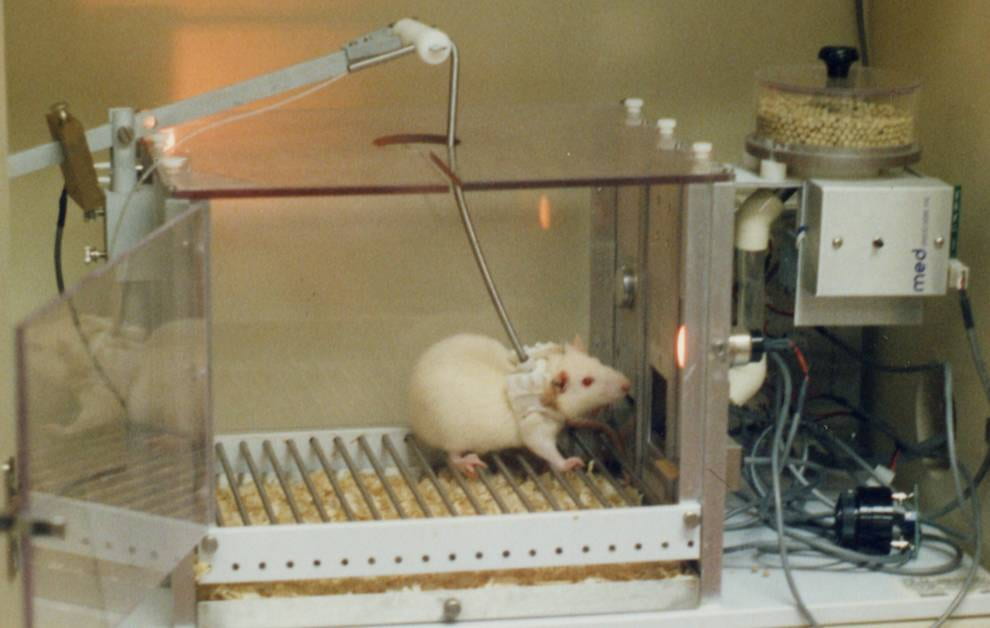
In Li et al’s research, mice were trained to self-administer cocaine by pressing a bar (see Fig 4 ). When pressed, cocaine solution was infused into their peritoneal cavity (the body cavity containing the stomach, intestines, liver etc) where it was absorbed into the blood. As found in many previous studies, once mice learned that bar pressing leads to cocaine administration, they readily and “enthusiastically” pressed the bar.
Twelve once-per-day sessions initially established the cocaine self-administration behavior. Once established, each cocaine self-administration was then paired with a painful foot shock. The mice responded to the paired shock by dividing themselves into 2 categories. Around 20% persisted in their cocaine administration despite the foot shocks (referred to as “persisters”) while 80% ceased administration (referred to as “renouncers”). Like human cocaine addicts, the persisters continued to self-administer despite negative consequences. It is interesting (although probably a coincidence) that the percentage of mouse persisters was similar to that of human cocaine users who become addicted.
Li et al determined that the difference between persisters and renouncers could not be explained by differences in learning the self-administration procedure, in how rewarding the cocaine was experienced, nor in how painful the foot shocks were perceived. These results indicated that something else must be influencing whether cocaine administration results in mouse “addiction.”
Optogenetic self stimulation of the reward pathway.
Electrically stimulating the ventral tegmental area, which also elevates dopamine in the nucleus accumbens, is both rewarding and addictive. However, the electrical stimulation quickly spreads out from the brain area being stimulated, preventing reliable neuroanatomical/neurochemical inferences. To get around this problem, Li et al took advantage of a newer technique called optogenetic stimulation. Optogenetic stimulation is not only better at confining stimulation to a localized area, it also provides precise millisecond control. In earlier research, Paoli et al (2018) found that mice will readily self stimulate by pressing a bar when the optogenetic stimulation is experienced as rewarding.
Optogenetic stimulation requires that the relevant neurons are infected with a virus that inserts a gene into the neuron’s genome that encodes a light-sensitive protein (a type of rhodopsin, similar to that which provides black and white vision in our eye). The rhodopsin embeds into the neuron’s cell membrane where it can be stimulated by a tiny optic fiber implanted into ventral tegmental area. When light is delivered through the optic fiber, the rhodopsin opens an ion channel that depolarizes the ventral segmental neurons causing dopamine release into the nucleus accumbens.
As with cocaine self administration, mice found this ontogenetic stimulation highly rewarding and readily learned to self stimulate by pressing a bar. After the mice were reliably self stimulating, a foot shock was paired with subsequent stimulations. As with cocaine self administration, some mice continued self administration (were “persisters”) while others became “renouncers” and quit.
However, an unexpected outcome was that optogenetic self-administration was even more “addictive” than cocaine self-administration (50% vs 12-20% persisters)! This difference turned out to be an important clue to understanding cocaine addiction liability in mice.
Why is optogenetic stimulation more addictive than cocaine stimulation?
An obvious difference is that cocaine went everywhere in the brain, while optogenetic stimulation was more confined to the reward pathway. As mentioned earlier, dopamine, serotonin, and norepinephrine concentrations were elevated in many brain areas by cocaine. Perhaps one of these 3 neurotransmitter was producing effects outside the reward circuitry that influenced cocaine addiction liability. Based upon prior research, serotonin was a likely possibility.
To test whether serotonin was involved, Li et al looked at cocaine self administration in two mouse strains (referred to here as normal and SertKI) that were genetically identical except for the gene encoding their serotonin reuptake transporter. In both strains, the serotonin transporter was effective at transporting extracellular serotonin back inside the neuron, however unlike the normal transporter, the SertKI transporter was not blocked by cocaine. Both the normal and SertKI mice experienced dopamine elevations in the nucleus accumbens following cocaine self administration and both readily self administered cocaine. However, unlike normal mice, the SertKI mice did not also experience serotonin elevations.
After establishing cocaine self administration, the cocaine was again paired with footshock. Li et al found that the SertKI mice were significantly more likely to become persisters than normal mice (56% vs 12%) with the percentage of SertKI persisters now resembling that of mice receiving optogenetic stimulation. The footshocks were similarly painful for both strains and could not account for the difference. Thus the elevated elevated extracellular serotonin caused by the cocaine appeared to suppress cocaine’s “addictiveness”.
However, one supportive experiment would not be sufficient to convince most scientists. Accordingly, Li et al performed 3 additional experiments from 2 additional perspectives to examine the possible role of serotonin. First of all, if optogenetic self stimulation is more addictive because it doesn’t elevate serotonin levels, then its addictiveness should be reduced by simultaneously elevating serotonin. To do so, citalopram, a SSRI antidepressant, was used to elevate serotonin levels throughout the brain during optogenetic self stimulation. Conversely, if cocaine is inhibiting its own addictiveness by elevating serotonin, then blocking the ability of the brain to respond to serotonin with either a serotonin receptor blocker (NAS181) or by using a strain lacking the relevant serotonin 2A receptor, should enhance cocaine’s addictiveness. Without going into detail, Li et al obtained the predicted results in all 3 experiments.
Consequently multiple lines of evidence provide convincing support that serotonin inhibits mouse “addiction” to both cocaine and optogenetic stimulation. The question then becomes, where in the brain does serotonin reduce addiction liability?
A neural pathway that influences addiction liability?

In earlier research, Pascoli et al identified a neural pathway that influences optogenetic self-stimulation under conditions of punishment. This pathway travels from the orbital frontal cortex (OFC) to the dorsal striatum (DS), and uses the neurotransmitter, glutamic acid (See figure 5). For brevity, this pathway will be referred to as the OFC/DS pathway. This pathway becomes active during optogenetic reward and becomes progressively “strengthened” over time by a process called long-term potentiation. (Long term potentiation is also thought to be a process by which conscious memories are permanently encoded in the cortex).
Long term potentiation results in both postsynaptic and presynaptic changes in glutamic acid synapses. The postsynaptic neuron becomes more sensitive to glutamic acid by increasing a type of glutamic acid receptor called an AMPA receptor. This increase makes it easier for glutamic acid to depolarize (i.e. activate) the postsynaptic neuron. Following repeated use, the presynaptic neuron also increases the amount of glutamic acid it releases each time it becomes active. These two changes result in the glutamic acid synapses becoming progressively easier and easier to activate. As the name “long term potentiation” implies, these changes are long lasting and can perhaps become permanent.
Very importantly, Pascoli et al also found that the activity in the OFC/DS pathway and the degree of strengthening was greater in the perseverers (i.e. addicted mice) than in the renouncers (i.e. unaddicted mice) providing evidence that this pathway plays a major role in extinguishing optogenetic self stimulation when accompanied by foot shock.
Li et al found that the OFC/DS pathway in mice self administering cocaine was similarly strengthened to a greater extent in perseverers than in renouncers. The remaining important issue was how serotonin inhibits the OFC/DS pathway.
How serotonin Inhibits the OFC/DS Pathway.
Li et al were able to figure out, at the cellular level, how serotonin decreases the likelihood of mouse addiction.
Virtually all serotonin neurons in the brain have their cell bodies localized in the Raphe nuclei in the brainstem. However, their axons project to many different areas in the brain and spinal cord. The dorsal striatum (DS) is one such area (See Figure 5). Although serotonin can work through traditional axodendritic or axosomatic synapses, Li et al found axoaxonic synapses to be most important for serotonin’s effect upon cocaine self administration in the DS. As seen in Figure 6, serotonin secreted across an axoaxonic synapse hyperpolarizes (inhibits) the glutamic acid terminal in the DS by binding to a serotonin 2A receptor which decreases glutamic acid release by OFC terminals. The inhibition of glutamic acid release also inhibits the development of long term potentiation which, in turn, decreases the likelihood of becoming a cocaine perseverer.

So why would the OFC and DS be involved in drug use and addiction? It is not surprising that the OFC would be involved in extinguishing addiction-related behaviors. The OFC is a part of prefrontal cortex, the executive center of the conscious brain. The prefrontal cortex makes conscious decisions about what you should be doing and when you should be doing it. Improper functioning of the prefrontal cortex often gives rise to poor decision making and poor impulse control, traits certainly seen in many drug addicts. (Google “Phineas Gage” for a fascinating example of how a damaged prefrontal cortex affects behavior ).
The DS consists mainly of two structures called the caudate nucleus and putamen, which are part of a more extended system called the basal ganglia. The basal ganglia are essential for helping motor cortex plan and execute motor movements and may also contribute to the motivation to perform these motor movements. Consequently, I would speculate that the parts of the DS activated by the OFC/DS pathway are providing guidance to motor cortex relevant to the execution of, and motivation for, addiction-related behaviors.
(As an aside, Parkinson’s disease is caused by a selective loss of dopamine input into the DS. As dopamine levels drop, the DS becomes progressively less able to provide necessary “advice” to motor cortex. As a result, the Parkinsonian patient progressively loses the ability both to initiate and terminate motor movements.)
Concluding remarks and implications for treatment.
First of all, some caveats. One should always be careful in generalizing mouse results to humans. While animal models of human processes are often relevant for understanding human processes, they should always be viewed with caution until verified by research with humans.
Even if relevant to humans, one should also be careful about generalizing the neurology of cocaine addiction to addiction to other drugs. At the same time, the plausibility is enhanced by the fact that two very different “addictive” procedures (cocaine self administration and optogenetic self stimulation) are both influenced by the same OFC/DS pathway in mice! Still, more research is necessary here as well.
I also left out details of much of the neuroanatomical research by both Pascoli et al and Li et al, that require a technical understanding beyond the scope of most readers (and to some extent, the writer of this post 😁). I do encourage those interested in a deeper understanding to read these 2 papers. While parts may be difficult to understand, these publications are excellent examples of the scientific method. Miyazaki and Miyazaki (2021) provide an easier-to-understand overview of the Li et al paper.
One question I have, is what sort of information does the prefrontal cortex, the executive part of the human brain, use in activating the OFC/DS pathway? The prefrontal cortex often integrates across a variety of inputs before arriving at decisions about what you should do. Obviously the reward pathway influences the prefrontal cortex’s decision to engage in drug use. In addition, input from the various senses, relevant cortical memories of past experiences, as well as unconscious parts of the forebrain including the hippocampus, amygdala, and basal ganglia are possibly involved as well. As I speculated earlier, perhaps even primitive parts of the unconscious brainstem contribute to the cortex’s decision to engage in drug-related behaviors.
Current addiction treatments are notoriously unsuccessful. Around 85% of addicts receiving treatment relapse within a year according to the National Institute of Drug Abuse. An important practical question raised by Li et al is whether cocaine addiction can be treated by methods that elevate brain serotonin. Perhaps the selective serotonin reuptake inhibitors (SSRI’s) used to treat depression might have efficacy. And would this treatment also work for other addictive drugs? If so, what would be the protocol for administering them? And would these findings be relevant only for preventing addiction, or also for reversing addition after it has been established? While both possibilities would be clinically significant, the latter is profoundly more important! Perhaps other methods of manipulating the OFC/DS pathway would have clinical relevance as well. Obviously these are all questions that require much future research.
So how might the Li et al paper be relevant to the question posed in this post’s title? Their findings suggest that overactivity of the OFC/DS pathway during drug use may be a predisposition to addiction. However this overactivity could arise from different neurological processes in different individuals. Perhaps in some individuals, lower levels of serotonin are available to inhibit this pathway. In other individuals, perhaps the problem is caused by a higher baseline of glutamic acid release. Alternatively, perhaps receptor sensitivity differences to serotonin or glutamic acid are the “culprits”. Or perhaps the problem might even be further “upstream” in the prefrontal cortex. One can envision other problematic causes as well. However, regardless of the “cause”, treating OFC/DS dysfunction might have efficacy in helping individuals better control their drug use.
Regardless, Li et al’s results provide potential new directions for better understanding human drug addiction and hopefully will provide new insights into treatment as well.
References.
Li, Y., Simmler, L. D., Van Zessen, R., Flakowski, J., Wan, J. X., Deng, F., . . . Luscher, C. (2021). Synaptic mechanism underlying serotonin modulation of transition to cocaine addiction. Science (New York, N.Y.), 373(6560), 1252-1256. doi:10.1126/science.abi9086 [doi]
Miyazaki, K., & Miyazaki KW. (2021). Increased serotonin prevents compulsion in addiction. Science (New York, N.Y.), 373(6560), 1197-1198. doi:10.1126/science.abl6285 [doi]
Pascoli, V., Hiver, A., Van Zessen, R., Loureiro, M., Achargui, R., Harada, M., . . . Luscher, C. (2018). Stochastic synaptic plasticity underlying compulsion in a model of addiction. Nature, 564(7736), 366-371. doi:10.1038/s41586-018-0789-4 [doi]



{kind=link}