Psychedelics have been used since before recorded history for both religious and medicinal purposes. However, their “discovery” by western medicine was relatively recent. A substantial body of evidence supports a variety of different psychedelic drugs having psychiatric value including glutamate psychedelics (eg. ketamine), serotonin-like psychedelics (including psilocybin, LSD and ayahuasca), and catecholamine psychedelics (eg. MDMA, aka Ecstasy). Except for ketamine, these psychedelics are currently DEA schedule 1 drugs (the same category as heroin) supposedly making them unavailable for medical purposes. However both psilocybin and MDMA were recently granted breakthrough status by the FDA for the purpose of allowing research to determine whether they are appropriate for therapeutic purposes. It seems likely to me that the FDA will, at some point, approve them for this purpose. This approval will also require the DEA to reschedule them. Ketamine, a DEA schedule 3 drug, is approved by the FDA as an anesthetic which then allows sub-anesthetic doses to be prescribed “off-label” to treat psychiatric issues such as depression and suicidal ideation.
Psychedelic Use in Psychiatry.
One of the earliest therapeutic uses of a psychedelic was a synthetic form of mescaline (a catecholamine-like psychedelic) as an adjunct to psychotherapy in the late 1930’s. However the first use of a serotonin-like psychedelic was likely that of LSD in the early 1940’s. LSD does not occur in nature and was first synthesized in the late 1930’s by Albert Hofmann, a Sandoz pharmacologist. However, its psychedelic properties weren’t discovered until 1943, when Hofmann accidentally dosed himself and had the first LSD “trip.” The potential implications for psychiatry quickly became apparent and shortly thereafter, Sandoz began making LSD available to psychiatrists and scientists under the brand name Delysid.
Over 2000 papers were published between 1960 and 1966 on the therapeutic uses of LSD! LSD was seen both as a model for understanding schizophrenia and as a treatment for a wide range of disorders, including depression, anxiety, schizophrenia, alcohol abuse, migraines, gambling and homosexuality. Therapeutically, it was used mainly for patients that did not respond to conventional treatments. Three different approaches were used: 1) Psycholytic therapy where LSD was used in conjunction with traditional Freudian psychoanalytic therapy; 2) Psychedelic Chemotherapy employing a more limited psychotherapeutic approach and 3) Psychedelic/Peak Therapy that attempted to induce a “mystical-like” experience. Evidence supported all three approaches having value for some disorders and all were considered safe in controlled medical settings. While LSD dominated this early phase of psychedelic research and therapy, other psychedelics such as mescaline and psilocybin were also used. Unfortunately this early work was performed before medical research began using modern research methods, leaving this work open to alternative explanations.
However, before modern research methods could be applied, these psychedelics were declared illegal. One factor leading to their illegality was the increasing recreational use of LSD in the 1960’s by “hippies,” whose counterculture perspectives many found threatening. “Turn on, tune in, and drop out” was a famous expression of the hippie movement popularized by Timothy Leary, a Harvard Professor and a leading proponent of recreational psychedelic use. As recreational use was not as controlled for dosage, mindset, and setting as medical use, the incidence of “bad trips” became more common. The resulting increase in emergency room visits and suicides following recreational use certainly contributed to the negative public opinion. In the 1960’s, there were also disturbing reports that the military had been trying to “weaponize” LSD as both a truth serum and as an incapacitating agent. There were also reports of the bad physiological effects of LSD, including chromosomal breakage, which were later proven incorrect.
By the mid 1960’s the damage to LSD’s reputation had been done and, in 1966 the Drug Abuse Control Amendments severely limited LSD sales and use. In response, Sandoz stopped making LSD, and LSD’s use in medical treatment and research was largely brought to a halt. And in 1970, the Comprehensive Drug Abuse Prevention and Control Act took it a step further and prohibited the therapeutic use of LSD and psilocybin altogether.
Public antipathy toward traditional psychedelics continued through the 1970s and 80s. This perspective was reinforced by Nixon’s “war on drugs.” Research proposing a link between LSD use and schizophrenia was also published at this time. At the same time, underground recreational and therapeutic use continued in the US and western Europe, with more open therapeutic use in Eastern Europe.
However, psychedelic therapy began making a comeback in the 1980’s, but with MDMA (commonly known as Ecstasy or Molly) rather than LSD. MDMA is a synthetic catecholamine-like psychedelic first synthesized in 1912 but not widely used as a psychotherapeutic drug until late 1970’s and early 1980s.
[As an aside, when MDMA began being used therapeutically in the 1980’s, it fell into a category that nowadays would be called “designer drug.” Until the DEA puts a new drug into one of its 5 schedules, there are no legal restrictions on its recreational or therapeutic use. However, a new drug cannot be scheduled until the DEA becomes aware of it and is able to study its effects. Unfortunately, this delay provides a window of opportunity for unscrupulous parties to develop and legally sell newly synthesized “designer drugs” before the DEA can schedule them.
While designer drugs sometimes work as advertised, they typically come with little or no toxicology or pharmacology. In some cases, particularly with designer opioids, the outcome has been disastrous. Despite the DEA making changes to attempt to close this “loophole”, the problem has only gotten worse. The most common designer drugs today are in the opioid, anabolic steroid, and psychedelic categories although there are also designer stimulants, sedatives, and viagra analogs as well as synthetic cannabinoids, synthetic cathinones and αMSH analogs (for tanning).]
By the late 1970’s, some psychiatrists were finding MDMA to be a more than adequate replacement for LSD. MDMA is not a classical serotonin-like psychedelic, but rather falls into the catecholamine-like category because its molecular structure closely resembles the catecholamines, norepinephrine and dopamine. The structure and effects of MDMA are similar to amphetamine, a drug that pharmacologists sometimes also place in the catecholamine-like psychedelic category. Amphetamine and MDMA both cause sympathetic arousal (i.e. “fight or flight” physiology) by increasing norepinephrine levels, are rewarding by increasing dopamine levels, and are psychedelic by binding serotonin 5-HT2A receptors. The differences between MDMA and amphetamine are largely a matter of degree. While amphetamine is more potent in stimulating sympathetic arousal, MDMA is more potent in stimulating psychedelic responses.
Psychotherapists found MDMA to have several desirable properties when compared to LSD. MDMA is shorter acting and produces a gentler, more rewarding, and more manageable psychedelic effect than LSD. Through promoting empathy and bonding, it appears to help patients work through emotional trauma and depression and appears particularly useful for treating Post Traumatic Stress Disorder (PTSD).
Although psychotherapists had hoped to confine MDMA to therapeutic use only, by the mid 1980’s it too was “discovered” by recreational users, most notably for its use at “rave” dance parties. While MDMA was considered safe in controlled medical settings, the same could not be said for its recreational use. The main problem was, when coupled with strenuous dancing, MDMA could cause hyperthermia and salt imbalance which could result in kidney and liver damage, and even death. Further adding to public disaffection was a published primate study showing that “recreational dosages” of MDMA caused brain damage. However, the paper was later shown to be incorrect and had to be retracted. In contrast, evidence from therapeutic settings did not provide evidence of long-term harm. Although there are risks associated with MDMA use, they are considered less serious than for legally available alcohol and tobacco. In 1985, the DEA nonetheless overruled input from medical professionals and placed MDMA in an emergency 1-year Schedule-1 category that prohibited both recreational and medical use. The scheduling was later made permanent.
In the 1990’s other psychedelics began to be explored for therapeutic potential, such as dimethyltriptamine (DMT), the active ingredient in ayahuasca, a psychedelic drug coming from plants mainly in S. America. There was also research using some of the newly synthesized catecholamine-like “designer” psychedelics before they could be scheduled by the DEA. However, underground therapy in the US and legal research elsewhere continued to be performed.
Ketamine was originally approved by the FDA in 1962 as an anesthetic. However around 2000, sub-anesthetic, psychedelic doses of ketamine were serendipitously found to possess significant antidepressant properties. It soon became apparent that ketamine was a major breakthrough for treating depressed patients who don’t respond to traditional antidepressants.Although approved by the FDA as an anesthetic, ketamine began being used“off label”as a treatment mainly for “treatment-resistant” depression.
In the early 2000’s the situation for the illegal psychedelics also began to turn around with the development of psychedelic research centers at prestigious universities, such as Johns Hopkins, and Harvard here in the U.S. and Imperial College London and Bristol University in the U.K. as well as funding by a number of organizations supporting psychedelic research. Some of the early research at these centers debunked many of the earlier negative findings. For example, when the concurrent use of other recreational drugs was accounted for, there was much less evidence for the serotonin-like psychedelics having severe negative effects. Although there are risks associated with MDMA use, they are considered less serious than for alcohol and tobacco.
And finally in 2018, as a step towards possibly legalizing therapeutic use, the FDA granted “breakthrough status” to psilocybin and MDMA. This status allowed medical professionals to treat depression and PTSD on an experimental basis. Unlike earlier treatments, these FDA-approved treatments are required to meet the exacting standards of FDA Phase II and III drug trials and are designed to examine therapeutic effectiveness under highly controlled conditions, determine the best treatment protocols, and also characterize any issues that might arise. Ultimately, this research could be used to determine whether FDA would approve these drugs for more widespread therapeutic use. So far the results look promising.
However, current views on the therapeutic use of psilocybin and MDMA by different organizations encompass the full range of possibilities. For example, the “official” position of the DEA, is that serotonin-like psychedelics (such as psilocybin) and catecholamine-like psychedelics (such as MDMA) are “hard drugs” whose extreme dangers outweigh any benefits (much like heroin, another schedule 1 drug) . On the other hand, many psychiatric experts (e.g. Reiff et al, 2020), as well as the FDA, believe these drugs have medical value but that more research is necessary before formal approval can be granted. At the other end of the spectrum, the state of Oregon recently “jumped the gun” and legalized psilocybin for therapeutic use by state-licensed professionals.
Some of these “illegal” psychedelic drugs will almost certainly receive federal approval for therapeutic use in the near future.Clearly, there are obvious parallels to the disjointed process by which cannabis (another DEA Schedule 1 drug) is being legalized here in the U.S.
References.
Reiff, C. M., Richman, E. E., Nemeroff, C. B., Carpenter, L. L., Widge, A. S., Rodriguez, C. I., . . . McDonald, W. M. (2020). Psychedelics and psychedelic-assisted psychotherapy. Ajp, 177(5), 391-410. doi:10.1176/appi.ajp.2019.19010035
Sessa, B. (2016). The history of psychedelics in medicine. In von Heyuden, M. et al (Ed.), Handbuch psychoaktive substanzen (pp. 1-26). Heidelberg: Springer -Verlag. doi:10.1007/978-3-642-55214-4_96-1
Tullis, P. (2021). The rise of psychedelic psychiatry. Nature, 589, 506-509.
In the 1940’s and 50’s, traditional serotonin-like psychedelics, such as LSD and psilocybin, were sometimes used to treat depression (and other disorders). In fact, some consider this early use to be a significant contribution to the beginnings of modern biological psychiatry. However, this use was derailed in the 1960’s when these drugs became illegal for any purpose (more about this in the next post). Currently, ketamine, a glutamate-related psychedelic, is the only psychedelic drug available for treating depression without special FDA permission.
Some psychiatric professionals strongly opposed the banning of psilocybin and LSD for therapeutic use, and, once banned, began advocating for overturning this restriction. Finally, in 2018 the FDA granted psilocybin “breakthrough status”, which permitted approved medical professionals to use it, on an experimental basis, for depression and other psychiatric disorders. This status was granted to expedite the evaluation of what the FDA now viewed as a “promising” therapy. Although psilocybin is currently the only traditional serotonin-like psychedelic with this status, other serotonin-like psychedelics, such as LSD and ayahuasca, would appear to possess similar antidepressant properties. In fact there is evidence that traditional psychedelics may be even more effective than ketamine.
In this post, background information is presented to put these drugs into a broader context and also lay some groundwork for future posts.
Different Classes of Psychedelics.
There are actually five different categories of psychedelic drugs defined either by their structural similarity to certain neurotransmitters, or, if they don’t physically resemble a neurotransmitter, by the type of neurotransmitter receptor(s) with which they interact. Traditional psychedelics, such as psilocybin, LSD, and ayahuasca, are all in the serotonin-like category because their molecular structure is similar to that of serotonin. Their psychedelic effects are also mediated through binding serotonin receptors. Ketamine, a glutamate-related psychedelic (also called a dissociative psychedelic), whose structure is different from glutamate, nonetheless works by binding a glutamate receptor.
But that’s not all folks! There are also catecholamine-like psychedelics (also referred to as empathogens or entactogens) whose structures resemble the catecholamines, norepinephrine and dopamine. While these psychedelics molecularly resemble catecholamine neurotransmitters, their psychedelic effect is actually caused by binding the same receptor(s) as the serotonin-related psychedelics. The final 2 classes are sometimes referred to as atypical psychedelics and include the acetylcholine-related psychedelics that work through binding an acetylcholine receptor, and an opioid-related psychedelic that works through binding an opiate receptor. There is evidence that catecholamine-like psychedelics such as MDMA, (also known as ecstasy) and the opioid-related psychedelic (salvinorin A) also have antidepressant properties.
I mention in passing that MDMA (also normally illegal for any purpose) also recently received FDA breakthrough status for treating post traumatic stress disorder (PTSD). Psilocybin also has efficacy in PTSD treatment, however, this and the next post will focus on the use of psilocybin and other serotonin-like psychedelics in the treatment of depression.
Serotonin-like Psychedelics.
The serotonin-like psychedelics are defined by their structural similarity to serotonin. As seen in Figure 1, they all contain the serotonin “carbon backbone” and, with the exception of LSD, differ differ mainly in possessing methyl (-CH3) functional groups. In fact most are essentially methylated versions of serotonin. Since serotonin itself is not psychedelic, the methyl groups have been proposed to confer the psychedelic and hallucinatory properties.
Figure 1: Serotonin and some serotonin-like psychedelics. The serotonin-like psychedelics are structurally similar to serotonin.
These psychedelics induce symptoms that bear some resemblance to the hallucinations and altered mental states seen in schizophrenia. In fact, the striking structural similarity of the serotonin-like and catecholamine-like psychedelics to neurotransmitters led to the “endogenous psychotogen hypothesis” of schizophrenia. According to this idea, schizophrenic symptoms are caused by abnormalities during neurotransmitter synthesis (such as methylation) resulting in defective neurotransmitters. While there are differences in the relative amounts of certain neurotransmitters in the brains of schizophrenics, there is no evidence that the neurotransmitter structures are abnormal. Consequently this hypothesis has not received much support.
At the same time, there is evidence that DMT (the psychedelic ingredient in ayahuasca) may be produced in small amounts by the pineal gland. However, there is not convincing evidence that endogenous DMT contributes to schizophrenic symptomology. And even if there were, most experts view schizophrenia as a complex disorder that likely has multiple causes.
It is worth noting that the serotonin-like psychedelics are not the only drugs that produce temporary schizophrenia-like symptoms. Glutamate-related psychedelics such as ketamine and phencyclidine can also produce such symptoms. In addition, chronic abuse of amphetamine, methamphetamine, or cocaine can also result in a temporary psychotic state that resembles schizophrenia. However, as yet, these “models” of schizophrenia have not provided definitive breakthroughs in our understanding of this complicated disorder.
And finally, in a small percentage of users, the serotonin-like psychedelics can precipitate a psychiatric disorder called Hallucinogen Persisting Perception Disorder (HPPD). In HPPD, hallucinations and other psychiatric symptoms continue to recur long after the drug has been cleared from the body. However, current thinking is that psychedelic drug use doesn’t so much cause this disorder as “reveal” it in predisposed individuals. For this reason, individuals with a history of psychosis, or evidence of a psychotic predisposition, are normally excluded from psychedelic therapies.
The Brain’s Serotonin System
Figure 2: The Serotonin System. The cell bodies of serotonin-releasing neurons are in the Raphe nucleus while their axons project throughout the brain and spinal cord.
Since serotonin-like psychedelics work through their interactions with serotonin receptors, a brief review of the brain’s serotonin system is in order. Serotonin is a neurotransmitter secreted by a small population of neurons whose cell bodies are in the Raphe Nuclei in the brainstem (see figure 2). However their unmyelinated axons project to virtually all areas of the brain and spinal cord. Upon reaching their destination, the terminals release serotonin both synaptically and extra-synaptically. These two modes of release complement each other. Synaptic transmission produces quick, punctate effects, while extra-synaptic volume transmission results in slower, more lasting, hormone-like effects.
Once released, serotonin can bind to, and activate, 15 different serotonin receptors that fall into 4 different gene families. These receptors are found in neuron membranes both inside and outside of synapses, and are also differentially expressed in different parts of the brain. Given this complexity, serotonin undoubtedly plays a variety of roles in different parts of the brain and spinal cord.
Fig 3. A schematic of serotonin secretion during wakefulness and sleep. The amount of serotonin secretion correlates with the amount of body movement. Secretion ceases altogether during REM sleep when the body is paralyzed. A brief burst of serotonin release at the beginning of a non-REM period is thought to delay the occurrence of the next REM period and contribute to the approximately 90-min periodicity of a REM + Non-REM bout.
Because serotonin secretion intensifies during physical exertion, one of its roles is thought to aid the brain and spinal cord in preparing for, and executing, motor movements. As would be expected, serotonin secretion shows a pronounced circadian rhythm, being highest when awake and physically active (See Figure 3). Daytime secretion declines as activity levels drop. Secretion declines even further after falling asleep and ceases altogether during Rapid Eye Movement (REM) Sleep when you are dreaming and your body is paralyzed.
As described in earlier posts, a chronic deficit in brain serotonin typically accompanies depression, while various treatments that boost brain serotonin are often effective in relieving depression. However, these serotonin-boosting treatments do not work for all patients indicating that brain serotonin concentration is only part of a more complicated story.
Mechanism of action of the serotonin-like psychedelics.
While the serotonin-like psychedelics also work through serotonin neurophysiology they do so quite differently from traditional antidepressants (such as SSRI’s). Whereas SSRI’s block serotonin reuptake transporters, serotonin-like psychedelics work as agonists for serotonin receptors.
However, these psychedelics do not bind all serotonin receptors, and typically bind with less effectiveness than serotonin (i.e. are partial agonists). One strategy for trying to understand which serotonin receptor(s) underly their psychedelic effect is to look for commonalities in the binding profiles of the different serotonin-like psychedelics. It turns out that all serotonin-like psychedelics show a high affinity for the serotonin 5-HT2A receptor (the chemical name for serotonin is 5-hydroxytryptophan, abbreviated 5-HT). The importance of this receptor is also supported by finding that the administration of a 5-HT2A receptor blocker (that blocks the ability of the psychedelic drug to bind) attenuates the psychedelic response. However there is also overlap in the binding of several other serotonin receptors, raising the possibility that other receptors may play a lesser role as well. Catecholamine-like psychedelics, such as MDMA (i.e. Ecstasy) also produce their psychedelic effect by binding the 5-HT2A receptor. One confusing relationship is that serotonin itself does not possess psychedelic properties despite binding the 5-HT2A receptor. So exactly how this receptor would confer psychedelic properties is unclear.
Psychedelic drugs often bind non-serotonin receptors as well. Although this binding may not contribute to psychedelic effects, it could possibly contribute to antidepressant properties (more about that when examining specific psychedelics) as well other side effects.
A Psychedelic “Trip”
Because taking a psychedelic drug is often perceived as going to a strangely different world, it is sometimes referred to as taking a trip. Certain aspects of this “trip” also appear central to the antidepressant effect. Since psychedelic experiences have been described in detail by others, I only briefly touch on them here, paying more attention to effects that might contribute to the treatment of depression.
These drugs are particularly well known for their perceptual distortions. Shortly after taking the drug, colors become brighter and geometric patterns can often be seen when closing the eyes (similar to “phosphenes” when rubbing your eyes). As the trip continues, these patterns intensify and can sometimes even be seen with eyes open, superimposed on the visual background. At this time, stationary objects may also seem to move or ripple or change texture and sometimes one object can morph into another. Auditory input is also altered and many report enhanced pleasure in listening to music. Taste and smell are also intensified and are also often experienced as more pleasurable. Sometimes the user may experience synesthesia, where sensory input seems cross circuited. For example, colors can be heard and sounds seen. Depending upon intensity, the sensory alterations can be classified as true hallucinations. Time also becomes distorted. Initially it appears to slow down but as the experience intensifies, time can appear to change speed, stop, or even go backwards.
Regular recreational users take psychedelics because they enjoy these bizarre effects, the intensity of which depend upon dosage and individual susceptibility. The time course of a trip typically varies from between 6 to 12 hours also depending upon dosage and susceptibility.
However, more relevant to its antidepressant effect, users can also have a “mystical” or “spiritual” experience. In this regard, these drugs are sometimes referred to as “entheogens” which means they release the divine within us. In such an experience, the normal boundaries that separate the individual from the the outside world appear to dissolve, resulting in a lessening of individual identity. This state is thought to permit the individual to look more realistically both “outward” and “inward”. When looking outward, they become more empathetic to, and understanding of, others. When introspectively looking inward, they are able to perceive themselves as other see them and in a more realistic fashion. In this state, the person is sometimes able to gain new perspectives on themselves and on their relationship to the world about them.
Some clinicians characterize this “ego-dissolving” mystical effect as a “peak experience” which, in turn, can lead to a restructuring of personality. In depressed individuals, this experience is hypothesized to reorganize brain processing in such a way that the patient no longer ruminates upon the negative mental processes underlying depression. A number of investigators provide evidence that such a psychedelic experience can have a lasting effect in relieving depression (several months and perhaps even permanently in some individuals). In this regard, several investigations have also noted a correlation between the intensity of the “mystical” experience and the magnitude of the antidepressant effect.
The downside is that not all users have positive experiences under the influence of these drugs. In fact, for some individuals, the experience can be terrifying and, in rare cases, even precipitate a psychotic episode. It has not been unusual for a recreational user experiencing high levels of anxiety and confusion to end up in a hospital emergency room seeking treatment. Obviously not an experience that you would want a depressed individual to undergo.
Whether an individual has a positive or negative experience is influenced by at least 3 variables. 1) The first is the mindset prior to taking the psychedelic. If the individual is convinced, in advance, that the experience will be positive, it usually is. 2) The setting in which the drug is taken is also important. A positive experience is optimized by taking the drug in a safe and comfortable environment where the user is free to experience the drug’s effects without outside interruptions or disturbances. It is also helpful to have an experienced person available who can provide guidance and reassurance if needed. 3) Negative and even psychotic reactions are most likely in individuals with a past history of psychosis or in individuals possessing such a predisposition (such as a person with a schizoid personality). Individuals known to possess such traits are generally excluded from psychedelic therapies. Since some hallucinogens can also cause a rise in blood pressure, individuals with severe cardiac disorders are also contraindicated. However, when appropriate measures are taken in controlled medical settings, few serious problems are normally observed.
Other related issues.
Drugs such as opiates, alcohol, nicotine, methamphetamine, cocaine, etc. are taken mainly because they activate the brain’s reward circuitry. They make you feel really good. Activation of the reward circuitry is central to understanding a drug’s abuse potential and addictive qualities. On the other hand, the serotonin-like psychedelics are generally poor activators of the brain’s reward circuitry. These psychedelics are taken mainly because users enjoy experiencing the perceptual distortions and altered states of consciousness. While the catecholamine-like psychedelics (such as ecstasy and other amphetamine-like drugs) are rewarding, they too are taken more for their perceptual and consciousness-altering properties than for their rewarding effects. Thus you typically do not see the level of abuse and addiction to psychedelics seen for other recreational drugs. At the same time, psychedelics do have their downsides. For example, the activation of the sympathetic nervous system by ecstasy (and other catecholaminergic psychedelics) can be fatal in rare cases, and all psychedelics can produce lingering psychiatric effects in a small percentage of users.
An issue I find particularly interesting, is why plants and fungi (and animals, in the case of toads) make psychedelics in the first place. It turns out that most psychedelic molecules possess one or more nitrogen atoms which classifies them as “alkaloids.” Alkaloids have 2 common properties: 1) they taste bitter and 2) they are often poisonous. In this way, organisms that make alkaloids protect themselves from being eaten. In many cases the bitter taste is sufficient. However, if that “warning” doesn’t work, many alkaloids also disrupt the functioning of the central nervous system. In some cases this disruption can be deadly (e.g. atropine, nicotine), but even if it isn’t deadly, negative consequences are likely. Insects, which are usually the greatest danger to plants, are generally more susceptible than humans and other vertebrates.
A related issue is that bitter taste perception in animals appears to have undergone parallel evolution to alkaloid evolution. The other 4 taste modalities (sweet, sour, salty and umami (savory)) have not changed much over evolutionary time and each can be accounted for by only one or a few genes. In contrast, there are 25 different bitter receptors in humans, each coded for by a different gene. In mice there are 35. The apparent reason is that, unlike other taste qualities, bitter tastants possess so many different chemical structures that more different receptors are required to detect them all. The number of bitter receptors in a given species appears related to the alkaloids regularly encountered by that species over evolutionary time. Clearly, bitter receptors have been a critically important animal adaptation to the evolution of plant (and toad) alkaloids.
And finally, I find it remarkable that we humans have exploited these “poisons” by turning them into drugs (e.g. nicotine, caffeine, heroin, morphine, cocaine, LSD, psilocybin, etc) that provide both recreational pleasure (at least sometimes) and medical therapy.
Concluding Remarks.
The next post will provide background on the history of the therapeutic use of traditional psychedelics.
Even though ketamine is considered by some to be the biggest antidepressant breakthrough in the past 50 years, an SSRI is almost always tried first. Sadly, SSRI’s work only about half the time on first attempt. Adjusting the dosage or switching to another monoamine antidepressant can sometimes address the problem. However, after failing two or more traditional antidepressant attempts, around 30% of depressed patients are designated “treatment resistant.” Ketamine is most often tried only after a patient receives this designation. (The various monoamine antidepressants are described in a previous post.)
Ketamine not only has an impressive 60-70% success rate in treatment-resistant patients, its antidepressant effect occurs almost immediately (versus 3-4 weeks for monoamine antidepressants)! Ketamine can also provide quick relief to acutely suicidal patients where its effectiveness is comparable to electroconvulsive shock (the gold standard for treating suicidality).
This post examines a number of issues relating to ketamine treatments of both depression and suicidality.
Why is ketamine a second-line antidepressant?
The first rigorous scientific demonstration of ketamine’s antidepressant effectiveness was in 2000. Since then, many studies have verified that ketamine is often effective when monoamine antidepressants are not. However, unlike monoamine antidepressants, ketamine treatment is “off-label.” Off label means that, while legal to use, the Food and Drug Administration (FDA) has not approved ketamine for this purpose.
Two questions that need addressing before proceeding are: 1) Since ketamine is more effective than the first-line SSRI’s, why isn’t ketamine tried first? (2) Since the scientific evidence strongly supports ketamine’s effectiveness, why hasn’t the FDA approved it as an antidepressant?
The major issue that prevents ketamine from being tried first is that ketamine is a DEA schedule-3 drug whose legal administration requires medical supervision. While ketamine is not expensive, its supervised administration is (more about cost below). In addition, these treatments need repeating on a regular basis since the antidepressant effects of ketamine are transitory. On the other hand, SSRI’s are much cheaper (even at full-cost, but often covered by insurance) and can be self-administered safely at home without medical supervision. So, if an SSRI is effective at treating depression and has manageable side effects, it is still considered the best option for most patients.
The reason ketamine is not FDA-approved for depression is that no one has sought approval. FDA approval requires extensive animal and human testing whose costs can sometimes exceed a billion dollars. Since the patent for ketamine expired long ago, it’s not worth the effort to go through this expensive process, only to be undercut by the availability of cheap generics.
Unfortunately, insurance companies often do not provide coverage for off-label drug treatments, particularly if expensive. Definitely a catch 22 for depressed patients who would benefit from, but can’t afford, ketamine treatments! In fact, since there is no commercial advertising for ketamine as an antidepressant, some insurance companies (and physicians) may not even be aware of this use.
Administration of Ketamine for Depression.
Before ketamine is administered, the patient typically is evaluated for suitability. As mentioned, most patients are in the treatment-resistant category. However ketamine is also appropriate for suicidal patients with a past history of depression or if a patient can not tolerate standard monoamine antidepressants. The patient must be judged healthy enough to receive ketamine by the standards of the American Society of Anesthesiologists. Contraindications can also include a past history of psychosis (including schizophrenia, schizoaffective disorder, or mania), hypersensitivity to ketamine, or prior drug abuse/addiction.
Ketamine is most often administered as a 0.5 mg/kg intravenous infusion over 40 minutes, although other routes, dosages, and time frames can also be effective. Administration normally occurs in a medical setting overseen by qualified professionals who monitor administration, acute-symptom recovery, and patient release. For experienced users, ketamine’s effects may be experienced as dreamlike, detached, relaxing, and rewarding. These positive effects also contribute to ketamine’s potential for abuse and addiction when used recreationally.
However, ketamine’s other acute effects can sometimes mimic experiencing a schizophrenic episode. These effects can be very unsettling and should be explained in advance. For example, ketamine can alter time and space perception and cause hallucinations in which you see and hear things that aren’t really there. Ketamine can also cause delusional thinking by impairing short-term memory and cognition. These psychotomimetic effects can sometimes trigger agitation and panic attacks. Since ketamine has analgesic properties, it also decreases the ability to feel pain which puts you at risk for hurting yourself without realizing it. Fortunately, these acute effects typically wear off in an hour or so. However, in psychiatrically predisposed individuals, ketamine can sometimes trigger a longer lasting psychiatric episode. Some patients feel that the most adverse psychological effect is a sense of dissociation, in which they feel strangely disconnected from themselves and the world about them. Paradoxically, in one study, the degree of dissociation was positively correlated with ketamine’s antidepressant effectiveness.
Other transient effects of ketamine can include a rise in blood pressure, nausea, vomiting, drowsiness, and dizziness. Vital signs are monitored throughout treatment and supportive care provided. However, these transient effects are well tolerated by most patients. Once the acute symptoms have worn off and vital signs have returned to normal, the patient can be released, usually within a few hours. However, patients are required not to drive or use heavy machinery for the rest of the day.
As ketamine’s acute effects wear off, most patients begin experiencing relief from depression almost immediately, with maximal effects around 24 hours later. This relief manifests as a reduction in negative thinking and in the obsessive negative spiral of depressive thought characteristic of depression. This relief is also described as an increased clarity of thought and as being different from the emotional smoothing caused by monoamine antidepressants. As with monoamine antidepressants, ketamine’s effectiveness in relation to a placebo is higher the more severe the depression. After a single treatment, ketamine’s antidepressant effect typically lasts about a week in unipolar depressed patients, although there can be substantial individual variation. While ketamine can also treat depression in depressed bipolar (manic/depressive) patients, it does not appear as effective. A single treatment for a bipolar patient usually loses effectiveness by day 3 or 4.
While IV infusion remains the most common method for treating depression, the other methods (subcutaneous, intramuscular, oral, sublingual, and intranasal) have been used successfully as well. Each has its own unique advantages and disadvantages in terms of ease of administration, dosage precision, first-pass metabolism, and absorption into the blood. While oral, nasal, and subcutaneous injections are convenient, these methods result in lesser, and more variable, absorption and also require higher dosages to achieve the desired result. Ketamine can also interact with other drugs that can alter ketamine’s effectiveness. For patients that do not respond initially, increasing the dosage, or administering repeated doses over a week or 2 can sometimes be effective.
In fact, repeated initial dosing is now the norm since it optimizes and prolongs effectiveness. Typical schedules might involve involve 4 treatments over a period of 1 or 2 weeks or 6 treatments over a period of 2 or 3 weeks. Sometimes the patient will be evaluated for their response to the first treatment as a basis for proceeding. However, all ketamine treatments must be administered by a qualified professional in a medical setting, making all treatments expensive. Most clinics charge between $350 – $800 per treatment. A full series of initial treatments can cost between $1000 – $12,000 and generally can be expected to last 2-3 weeks.
Since ketamine’s antidepressant effects are temporary, the patient will almost certainly need re-treatment. Ideally re-treatment should occur before the previous treatment has worn off. Since this time frame can vary from person to person, trial and error may be required. A single re-treatment every week or so is often sufficient to maintain the antidepressant effect and some patients have now been treated successfully for years. The acute side effects of ketamine also seem to diminish with re-treatment, however, there has not been rigorous research on long-term treatment on overall health which provides concerns.
There may be the possibility of unsupervised ketamine self-administration in the future which almost certainly lower costs and increase ketamine’s antidepressant use. Andrade (2019) described 3 studies to examine this possibility that involved drinking a ketamine solution. Once successful dosage and re-treatment schedules were clearly established under professional supervision, patients were able to successfully self-administer and treat their depression at home. The most common side-effects were light-headedness, sedation, and mild dissociative symptoms which typically subsided within an hour. Taking ketamine just before going to bed seemed to minimize these symptoms. In fact, Andrade (2019) makes the argument that, when the ketamine solution was sipped over a 10-15 minute period, the slow absorption into the blood made this method even safer than IV administration. Ketamine does taste very bad, but its taste can be masked by flavoring agents. A company called Mindbloom is attempting to make ketamine self administration at home possible, although expert’s are not in agreement as to whether we know enough for this to be a good idea.
While ketamine can help many treatment-resistant patients, its cost and typical methods of administration prevent it from being ideal. In addition, around 30% of treatment-resistant patients do not respond therapeutically to ketamine. Other strategies are necessary to try to help these patients.
(Since writing this post 3 years ago, it has become possible to get prescriptions for ketamine through telemedicine. While this is definitely helpful to many depressed patients it is also leading to serious misuse/abuse!)
Ketamine’s effects on other correlates of depression.
As with other successful depression treatments, effective ketamine treatment also normalizes other symptoms accompanying depression. For example sleep disturbances and biological rhythms are improved as are cognitive/memory issues. With regard to sleep, ketamine treatment restores the normal levels of slow-wave sleep most prevalent early in the night. One reason this is important is that slow-wave sleep corresponds to the time when the brain is maximally “cleansing itself” by exporting toxic metabolites into general circulation for removal. While the acute effects of ketamine disrupt cognitive processes, it is interesting that the longer term effects are the opposite. Impairments of general health such as metabolic syndrome and enhanced inflammation are also reduced in ketamine-treated patients. And finally, as with other successful antidepressant treatments, ketamine enhances synaptic plasticity and repair of limbic system and cortical abnormalities.
Ketamine treatment of suicidality.
Around 800,000 people worldwide die from suicide each year and many more harm themselves in unsuccessful attempts. Moreover, each unsuccessful attempt increases the likelihood of a subsequent attempt. Effective, accessible treatments are greatly needed.
All suicide treatments have limitations. Traditional monamine antidepressants can reduce suicidal tendencies in some patients. However, since these treatments require 3 or more weeks for effectiveness, they are not very useful for acutely suicidal patients. Electroconvulsive treatment (ECT) historically has been the gold standard for treating suicidally depressed individuals. Compared to monoamine antidepressants, ECT is both quicker acting and more effective. However, because of the specialized equipment and expertise required, access is limited, and wait lists can be long. ECT also has the downsides of being costly and causing some memory loss. And finally it’s mischaracterization in movies and the popular press has created an enduring stigma that, no doubt, reduces its use.
Nowadays, ketamine is being used off label as an alternative to ECT. Ketamine’s effectiveness appears comparable and its speed of action may actually be quicker. And, unlike ECT, virtually all hospitals are equipped to administer ketamine. For some patients a single ketamine treatment may be sufficient although multiple treatments over several days are more typical. Although IV infusion is the most common method of administration, oral, nasal, and IM administration make it even more convenient.
Case studies of suicidal unipolar and bipolar patients have, in many cases, demonstrated rapid and profound effectiveness in reducing the hopelessness that often underlies suicidality. However, some patients require multiple treatments over several days. Experimental support also comes from suicide rating scales administered both before and after ketamine treatment. Whether ketamine would be effective for suicidality associated with other psychiatric disorders such as schizophrenia or obsessive/compulsive disorder has not been conclusively determined.
However, as is the case for depression, ketamine’s anti-suicidal effects are transitory, a single treatment typically lasts about a week. As with depression, daily treatments over several days can prolong effectiveness. Although it seems likely that ketamine maintenance therapy can prevent recurrence, limited evidence is available. Although there are many parallels between ketamine’s effect upon depression and suicidality, there is some disagreement as to whether ketamine’s anti-suicidal effect is the same as its antidepressant effect. This argument seems to hinge, in part, on whether ketamine is effective for non-depressed suicidal patients.
In general, the research on ketamine and suicidality is much less developed than the research on depression. Research is currently underway to compare ketamine and ECT (the two most effective treatments), both separately and in combination, in hopes of optimizing future treatments.
S-ketamine (Esketamine) vs R-ketamine.
As described in the previous post, after synthesis, racemic ketamine is comprised of equal amounts of 2 mirror-image molecules called S-ketamine and R-ketamine. Although complicated to do so, the 2 molecules can be chemically separated. Of the two, S-ketamine is the more potent antagonist of the NMDA receptor, resulting in better anesthesia/analgesia while also causing less drowsiness and cognitive impairment. Randomized, double blind, placebo-controlled research demonstrates that S-ketamine is also an effective antidepressant when administered either IV or intranasally, either by itself or in combination with monoamine antidepressants.
In March of 2019, the S-enantiomer (also called esketamine and patented as Spravato by Janssen Pharmaceuticals), became only the second drug approved by the FDA for treatment-resistant depression. Spravato received a FDA fast-track designation because of the great need for approved treatments for treatment-resistant patients. Spravato also received FDA approval for suicidially depressed patients.
While Spravato treatment is expensive (first month costs are estimated to be between $4000 and $6000), the out-of-pocket costs can be much lower because of insurance coverage (although some insurance companies might be reluctant to cover these costs). To Spravato’s advantage, the only other FDA-approved drug for treatment-resistant depression, Symbyax, is much slower acting, often requiring a month of treatment to be effective. (Symbyax is a combination of fluoxetine, an SSRI; and olanzapine, an atypical antipsychotic used for treating schizophrenia.) While Janssen is predicting blockbuster sales of Spravato, cost and accessibility may be issues for many patients.
To meet FDA requirements, Spravato is administered as a nasal spray and is required to be combined with a traditional oral monoamine antidepressant (typically an SSRI). While nasal administration is more convenient than IV infusion, Spravato’s administration still requires medical supervision. Although Spravato can be used off label for other purposes, it’s not clear that there would be much advantage over standard ketamine treatments.
While S-ketamine is clearly a more potent anesthetic/analgesic than R-ketamine, it’s not clear that S-ketamine is the more effective antidepressant. In fact, in animal models of depression, R-ketamine had longer lasting antidepressant effects with fewer adverse psychotomimetic side effects. R-ketamine also was better at enhancing the neuroplasticity that corrects depression-related neuropathologies. Perception Pharmaceuticals is currently investigating R-ketamine for antidepressant use in humans. However, rigorous comparisons in humans of the antidepressant properties of two enantiomers and the racemic mixture have not been performed.
Other Ketamine-like Drugs?
To displace the SSRI’s as a first-line antidepressant, a new drug would not only have to be more effective, it would also have to be safe enough for self administration without medical supervision. Attempts are underway to develop new antidepressants that have ketamine’s antidepressant property without its acute psychotomimetic side effects. Whether this approach can ultimately produce first-line antidepressants, or just better second-line antidepressants isn’t clear.
Ketamine is thought by some to produce both its antidepressant and its psychedelic effects by completely blocking ion flow through the NMDA ion channel. One strategy for developing new antidepressants would be to only partially block ion flow. The idea is that the reduced ion flow would be sufficient to provide the antidepressant effect, but insufficient to trigger the undesired psychotomimetic side effects.
One approach using this reasoning depends upon the fact that the NMDA receptor has binding sites for other molecules as well. The molecules binding these alternative sites are referred to as allosteric modulators. When bound, these modulators very slightly alter the 3-D shape of the NMDA receptor which, in turn, either increases or decreases the ability of glutamate to open the NMDA ion channel.
Figure 1: Schematic of an NMDA receptor. Glutamate binding is necessary to open the ion channel. However a precondition is that glycine must first be attached to its binding site. Glycine binding is inhibited by agonists of the GlyX binding site. However partial GlyX agonists should only partially block ion flow. (click on graphic to enlarge)
As seen in Figure 1, glycine is one such allosteric modulator whose binding is a necessary precondition for glutamate to open the ion channel. To make matters more complicated, there is another binding site, termed the GlyX site, separate from the glycine site, that modulates the ability of glycine to bind the glycine binding site. When a GlyX agonist binds the GlyX binding site, it blocks glycine binding. On the other hand, a partial GlyX agonist only partially blocks glycine binding. Thus a partial agonist should cause the desired partial reduction in glutamate-activated ion flow.
Joseph Moskal of Allergan Pharmaceuticals has, in fact, developed a partial agonist for the GlyX binding site, originally given the code name GlyX13 (and later the brand name of Repastinel). In animal testing, Repastinel had rapid antidepressant effects similar to ketamine, but without ketamine’s pronounced psychotomimetic side effects. As a result the drug was fast tracked through FDA testing. Unfortunately, animal results don’t always translate to humans, and in FDA Phase III human trials in 2019, Repastinel was not better than a placebo as an antidepressant. As a result, further development of Repastinel was discontinued.
However, Allergan has not given up on this approach and currently has another drug in development that also works through the GlyX binding site (code name NRX-1074, also known as Apimostinel) . Apimostinel differs from Repastinel in being a more potent partial agonist and can be administered either IV and orally, in contrast to Repastinel which could only be administered IV. Preliminary animal testing was consistent with antidepressant action without psychotomimetic side effects. Hopefully this drug will succeed in human trials.
Several other pharmaceutical companies have similar drugs under development, targeting both NMDA receptors as well as other types of glutamate receptors in hopes of developing better antidepressants. While one of these antidepressant candidates may prove effective and have fewer side effects than ketamine, most will likely require medical supervision and target mainly treatment-resistant patients unresponsive to monoamine antidepressants. While these new drugs will be expensive, the good news is that with FDA approval, insurance coverage becomes more likely.
A Glutaminergic Model of Depression?
Abnormal glutamate functioning in the etiology of depression is certainly evidenced by excessive glutamate neurosecretion and a decrease in glutamate synapse formation in depressed individuals, as well as by ketamine’s effectiveness in correcting these abnormalities. In addition the brain areas most implicated in depression (the limbic system and cortex) are heavily dependent upon the functioning of glutamate-secreting neurons. In fact, neuroimaging indicates these brain areas are preferentially targeted by ketamine. Consequently there is good reason for thinking that glutamate malfunctioning may be central to understanding depression. However, this post is already too long 🥱 ……… so a post for another day.
Next Post.
The next post will look at another class of psychedelic drugs (including psilocybin, LSD, and ayahuasca), that also appear more effective than than the current first-line SSRI antidepressants.
Addendum: The effect of telemedicine on ketamine administration for depression.
Since posting this article a few years ago, the situation has changed for the use of ketamine in treating depression (and other psychiatric issues). During the COVID Epidemic, in order to make prescription medicines more accessible and less expensive, the rules were changed to allow for telemedicine prescriptions. These changes were initiated during the Trump administration but have been continued by the Biden adminitration, After a telephone or video interview with a remote physician, the prescription can be issued, filled online, and delivered by mail. An oral (rather than injectable) version of ketamine is often tailored to the patient’s prescription by a compounding pharmacy. This change allows ketamine to be self-administered at home by the patient. This change has, no doubt, benefited many depressed patients who take ketamine according to directions.
However, telemedicine also has a downside. As pointed out in the New York Times, this procedure makes it possible for a patient to abuse their prescription by taking more than recommended. The risk is likely enhanced by a psychiatric condition. The long-term effects of ketamine abuse are not fully known, however it is clear that some patients become addicted and some suffer bladder-control problems. Perhaps some form of patient monitoring is needed for ketamine (and other potentially dangerous drugs).
Some articles for further reading.
The reviews by Andrade cover the full range of issues relevant to ketamine’s antidepressant use and are intended mainly for educating clinicians. The other reviews are written for a more technical scientific audience. Much of the information presented in this post can be found in these reviews.
Andrade, C. (2017a). Ketamine for depression, 1: Clinical summary of issues related to efficacy, adverse effects, and mechanism of action. The Journal of Clinical Psychiatry, 78(4), e415-e419. doi:10.4088/JCP.17f11567 [doi]
Andrade, C. (2017b). Ketamine for depression, 2: Diagnostic and contextual indications. The Journal of Clinical Psychiatry, 78(5), e555-e558. doi:10.4088/JCP.17f11629 [doi]
Andrade, C. (2017c). Ketamine for depression, 3: Does chirality matter? The Journal of Clinical Psychiatry, 78(6), e674-e677. doi:10.4088/JCP.17f11681 [doi]
Andrade, C. (2017d). Ketamine for depression, 4: In what dose, at what rate, by what route, for how long, and at what frequency? The Journal of Clinical Psychiatry, 78(7), e852-e857. doi:10.4088/JCP.17f11738 [doi]
Andrade, C. (2017e). Ketamine for depression, 5: Potential pharmacokinetic and pharmacodynamic drug interactions. The Journal of Clinical Psychiatry, 78(7), e858-e861. doi:10.4088/JCP.17f11802 [doi]
Andrade, C. (2019). Oral ketamine for depression. Journal of Clinical Psychiatry, 80(2), e1-e5.
Corriger, A., & Pickering, G. (2019). Ketamine and depression: A narrative review. Drug Design, Development and Therapy, 13, 3051-3067. doi:10.2147/DDDT.S221437 [doi]
Matveychuk, D., Thomas, R. K., Swainson, J., Khullar, A., MacKay, M. A., Baker, G. B., & Dursun, S. M. (2020). Ketamine as an antidepressant: Overview of its mechanisms of action and potential predictive biomarkers. Therapeutic Advances in Psychopharmacology, 10, 2045125320916657. doi:10.1177/2045125320916657 [doi]
Muller, J., Pentyala, S., Dilger, J., & Pentyala, S. (2016). Ketamine enantiomers in the rapid and sustained antidepressant effects. Therapeutic Advances in Psychopharmacology, 6(3), 185-192. doi:10.1177/2045125316631267 [doi]
Ketamine is thought by some to be the biggest breakthrough in the treatment of depression in the last 50 years. However to provide broader perspective on ketamine’s use as an antidepressant, this post looks at its original role as an anesthetic and why it can also be a recreational drug of abuse. In addition, this post looks at ketamine’s mechanism of anesthetic action, the 2 different ketamine variants, and the various ways ketamine can be administered. The next post will look at ketamine’s role as an antidepressant.
Ketamine as an anesthetic.
Ketamine is a relatively short-acting synthetic drug, FDA-approved as an anesthetic in 1970. Ketamine’s action is terminated by liver enzymes that degrade it into metabolites that are excreted, mainly in the urine. At the appropriate dosage, ketamine has the anesthetic properties of rendering a patient both unconscious and amnestic to events while anesthetized. At the same time, ketamine has some other desirable characteristics that distinguish it from most other anesthetics. These include an unusually good safety profile; little respiratory or circulatory depression; and analgesia, reducing the need for pain medication.
At the same time, ketamine has its downsides. When fully anesthetized, the patient strangely appears as if they might be awake, with their eyes open and with noticeable muscle tone. Since some body movement is possible, ketamine is less desirable when movement is detrimental to medical procedures. In addition, ketamine has the same mode of action as phencyclidine, an anesthetic drug removed from the market in 1955 because it can produce a temporary, dissociative, trance-like, catatonic psychosis indistinguishable for schizophrenia. Ketamine can also produce these symptoms but, because it is less potent and shorter acting, the effects are typically less severe. Around 10-20% of patients experience hallucinations and delusions upon emerging from ketamine anesthesia, although the effects usually wear off quickly without lasting effects. However, ketamine can cause more prolonged psychiatric symptoms in psychiatrically predisposed individuals. Interestingly, this psychotomimetic effect is more pronounced in adults than children and becomes more likely to occur after early-adulthood, the time when schizophrenic symptoms are typically first noticed. (In fact, the phencyclidine/ketamine “psychosis” has contributed to our understanding of the neurological underpinnings of schizophrenia). Ketamine’s potential psychiatric side effect certainly provides a caution for its use.
An additional downside is that at the subanesthetic doses used for treating depression, ketamine is rewarding and potentially addictive. This effect, in part, underlies its illicit recreational use as a club drug (some street names: K, Special K, Super K, Vitamin K, Donkey Dust, Cat Valium, Ket, and Wonk). However, ketamine is also used recreationally for its hallucinogenic and dissociative properties. Unfortunately, chronic abuse can lead to liver and kidney toxicity. Ketamine can also be used as a date rape drug. Historically, ketamine’s illicit uses have been diverted mainly from veterinary supplies.
Nonetheless, because ketamine’s desirable properties sometimes outweigh its downsides, it remains a valuable anesthetic. For example, because of its safety and reduced need for accompanying analgesia, ketamine was used extensively as an emergency field anesthetic during the Vietnam war. Nowadays ketamine is used as a pediatric anesthetic since children are unlikely to experience psychiatric side-effects. Because ketamine doesn’t depress breathing, it is also used with asthmatics, individuals suffering from obstructive airway issues, or if ventilation equipment is not available. Ketamine is also sometimes used as a preanesthetic to prepare patients for surgery which allows its psychoactive effects to wear off by the time the patient awakens, and also sometimes for its analgesic properties. Ketamine is used even more extensively as a first-line veterinary anesthetic.
Ketamine anesthesia works by binding the NMDA receptor.
Ketamine is a pharmacologically “messy” drug that binds numerous receptors in the brain. However, ketamine’s highest affinity is for the N-methyl-D-Aspartate (NMDA) receptor, a type of glutamic acid (i.e glutamate) receptor, which mediates its anesthetic, analgesic, and amnestic effects. As seen in figure 1, The NMDA receptor is an ionotropic receptor in which 4 proteins join together in the cell membrane to provide both an extracellular glutamate binding site as well as an ion channel through the membrane. The 4 proteins are of 2 types: R1 and R2. In addition, several different genes code for the different subtypes of the R2 protein resulting in a variety of ways of assembling the NMDA receptor. The interchangeable R2 subtypes, at least in part, provide redundancy so that if one gene is defective, functional receptors can still be formed. In addition, differential R2 gene expression in different parts of the brain might also serve to optimize local NMDA functioning. It is worth noting that biological systems possessing redundant “backup systems” are generally those most crucial to survival.
Figure 1: Schematic representation of an NMDA receptor showing the binding sites for glutamate and ketamine as well as other molecules that can modulate the ability of glutamate to open the ion channel. Double click on graphic to enlarge.
In the receptor’s resting state, the relatively nonselective ion channel (seen in blue) is closed and requires glutamate binding to open. However, several preconditions must first be met including that the membrane be depolarized and that glycine be attached to its binding site. The ability of glutamate to open the ion channel can also be modulated by other molecules such as magnesium, zinc, and ethanol attaching to their respective binding sites as seen in Figure 1.
Once preconditions are met, glutamate binding opens the ion channel and 4 of the small ions in biological fluids (Ca++, Na+, K+, and Cl–) are free to move down their concentration gradients, through the ion channel, and across the membrane. Ca++ and Na+ are more prevalent in the extracellular fluid, so they move to the inside of the cell, while K+ and Cl–, more prevalent in the cytoplasm, do the opposite. The electrical charges of the ions crossing the membrane come close to cancelling each other out and make only a negligible contribution to neuron excitability. However the entry of Ca++ is critical for activating intracellular enzymes underlying the brain’s capacity to form new glutamate synapses as well as strengthening existing ones. This “neuroplasticity” is incredibly important as it provides the physical basis for our capacities for learning, memory, and ultimately cognition! As a result, the NMDA receptor is among the most studied receptors in the brain.
Ketamine exerts its effects by attaching to its binding site inside the ion channel (seen in Figure 1), physically blocking the channel and preventing glutamate’s ability to initiate ion flow. The immediate effects are very disruptive to brain functioning and cause ketamine’s anesthetic, analgesic, amnestic, and psychotomimetic effects. However, after these immediate effects have subsided, ketamine’s longer term effects somehow reduce depression symptoms even more effectively than the first-line SSRI antidepressants! More about that in the next post.
Different ketamine enantiomers.
Figure 2: The two mirror-image enantiomers of ketamine.
Ketamine is synthesized in pharmaceutical laboratories as a “racemic” mixture consisting of equal amounts of two chemically identical, but spatially different molecules (called “enantiomers”), termed R-ketamine and S-ketamine. The binding of these mirror-image molecules to brain receptors is analogous to putting your hands into a glove. Although either hand can be put into either glove, the right hand fits best in the right glove and the left in the left. The same is true for these 2 enantiomers, each fits certain brain binding sites better than the other.
Once the racemic mixture is synthesized, it is possible to chemically separate the two enantiomers, although the process is both difficult and expensive. The S-enantiomer (also called esketamine) is the more potent anesthetic and analgesic because it more effectively blocks ion flow through the NMDA ion channel. Other differences from R-ketamine are that the S-ketamine is cleared from the body quicker, produces less impairment of cognition, less loss of concentration, fewer psychotic reactions and less agitated behavior.
The S-version recently recently received FDA approval as an antidepressant under certain conditions (more about that in the next post). However, the racemic mixture containing both enantiomers remains the most common formulation for both anesthetic and antidepressant use.
How is ketamine is administered?
Ketamine is available as a white powder or as an aqueous solution and can be administered intravenously, intramuscularly, subcutaneously, orally, rectally or intranasally. These methods differ significantly in first-pass metabolism and in percentage absorption into the blood. First-pass metabolism (from enzymes in the digestive system and liver) intervenes between drug administration and entry into general circulation and contributes to differences in bioavailability (percentage of administered drug that actually gets into the blood). First-pass metabolism also introduces ketamine metabolites into general circulation, some of which also have anesthetic and antidepressant properties. This contribution has not been well studied in humans and could have implications for dosage.
For depression, ketamine is most often administered as an aqueous solution via intravenous (IV) infusion over a period of around 40 minutes. Unlike the other methods, IV administration results in 100% bioavailability and no first-pass metabolism which allows for precise dosage control. The slow rate of IV infusion is also thought to minimize some of ketamine’s acute side effects. An additional advantage is that the dosage can be adjusted during the course of administration. The other methods can also be effective, but do not allow for such dosage adjustments and are also less predictable because of individual variability in first-pass metabolism and absorption. There is no antidote for ketamine toxicity, however, all methods are generally considered safe in the dosages used for treating depression.
Next Post.
The next post looks at ketamine’s role as an antidepressant
The traditional pharmacological methods of treating depression leave a lot to be desired.The monoamine antidepressants, which include the first-line selective serotonin reuptake inhibitors (SSRI’s), end up working for just 70% of depressed patients. However only 50% respond on the first attempt. For those not initially responding, dosage adjustments or perhaps switching to another antidepressant can sometimes help. However a treatment can take about a month to determine effectiveness and if multiple attempts are necessary, even more time will be required.Furthermore, even when “effective”, the therapeutic outcome can be less than desired.Clearly we need quicker acting, more effective, first-line antidepressants .
While the first-line SSRI’s have fewer side effects than the earlier generation antidepressants, side effects can still be significant.Side effects can include headaches, nausea, trouble sleeping, dizziness, diarrhea, fatigue, anxiety, stomach upset, dry mouth, and sexual problems such as low sex drive, erectile dysfunction or ejaculation problems. While these side effects often diminish over time, they nonetheless make compliance difficult for some patients. Overdosing a patient can also cause serotonin syndrome (described in an earlier post under “SSRI side effects”) which, in extreme cases, requires hospitalization.
Yet another problematic issue with monamine antidepressants is that when taken over an extended period, patients can develop pharmacological tolerance.If a patient then discontinues treatment, unpleasant withdrawal symptoms can sometimes last a month or more (also referred to as antidepressant discontinuation syndrome).Although withdrawal symptoms can be diminished by gradually tapering the drug and taking other medications to counteract withdrawal symptoms, some patients experience sufficiently unpleasant symptoms that they choose not to quit.
The 30% of patients who do not respond to 2 or more standard antidepressant treatments are termed “treatment resistant.”There are “last-resort”, non-drug therapies that can help.These treatments include electroconvulsive-shock therapy (ECT), repetitive transcranial magnetic stimulation, vagus nerve stimulation, and deep-brain stimulation. Of these, ECT is the most used and most effective, and by some accounts, even more effective than traditional antidepressant drugs.However, in addition to invasiveness, these treatments are expensive because they require hospital settings, specialized equipment, and a team of trained professionals.For example, each ECT session costs about $2,500 and the typical 10 or so sessions over a period of several weeks would cost around $25,000, plus any additional costs of a hospital stay.Depending upon one’s insurance, these costs may, or may not, be covered. There are also no guarantees of lasting effects.
The good news is that we now have two classes of “antidepressant” psychedelic drugs that are both quicker and more effective than the monoamine antidepressants. The bad news is that their high cost and limited availability puts them out of reach for many individuals. The drugs themselves are not particularly expensive. However, like the non-drug therapies, the cost of treatment is. Because of their federal classifications, these drugs must be administered under licensed medical supervision. Self administration outside of medical settings, for either therapeutic or recreational purposes, is illegal.
One experimental class of psychedelics (including LSD, psilocybin, and ayahuasca) has limited availability for treating depression. Because these drugs are not normally allowed for medical use, each therapist in the USA must obtain special FDA approval. However, the other class (including ketamine and its derivatives) can more routinely be used “off label”. Off label means that while the drug is available for medical use, the FDA has not given formal approval for its use as an antidepressant. Because medical use is off label, insurance companies typically do not cover costs, which can run into thousands of dollars per month.
There is hope that scientists can discover new drugs that retain the antidepressant effect of psychedelics without their acute psychoactive effects. Unless that happens these psychedelics are likely to remain secondary antidepressants, used mainly for treatment-resistant patients as an alternative to the more invasive non-drug therapies.
The remaining posts on depression explore the use of these psychedelics for treating depression.However to provide background, other medical and recreational uses of these drugs are explored as well.
The monoamine antidepressants were the first effective drugs to treat depression. Because they work by boosting the brain’s monoamine neurotransmitter concentrations, this led scientists to hypothesize that depression is caused by insufficient monoamine activity.
In this post, I explain how monoamines are released by axon terminals, how they interact with their receptors, and how these relationships are thought to relate to depression. I then present a brief overview of the different classes of monoamine antidepressants, their side-effects, and how monoamine antidepressants have (and haven’t) changed over the years.
Synaptic Transmission vs Volume Transmission
Synaptic transmission and volume transmission are two different ways that axon terminals release neurotransmitters (See Figure 1). Release across a synapse involves the axon terminal being very close to its receptors, while in volume transmission the axon terminal is more distant. The pattern of neurotransmitter release is also different. In synaptic transmission, release can be highly variable where the pattern and amount of release encode specific information. In contrast, volume transmission involves more regular release whose amount changes more slowly.
Since volume transmission results in monoamines being released farther away from receptors and into a larger volume of extracellular fluid, the peak monoamine concentration at the receptor is typically lower than in synapses. Also neurotransmitters released by volume transmission are more difficult to remove from the extracellular fluid by reuptake (see figure 2) since the monoamine molecules are often farther away from the reuptake transporters that remove them.
Figure 1. Synaptic vs Volume Transmission. Synaptic transmission involves the release of a neurotransmitter across a synapse that interacts almost exclusively with receptors in the postsynaptic membrane. Volume transmission involves release directly into the extracellular fluid further away from its receptors and can interact with receptors both inside and outside of synapses.
While the concentration of the neurotransmitter in a synapse can change dramatically in milliseconds, outside the synapse the monoamine concentration from volume transmission changes more slowly. However, the concentration outside of synapses does change under certain conditions. In the case of serotonin, concentrations increase just before body movements, which is thought to hep prepare the brain and spinal cord to contribute to movement. Norepinephrine concentrations increase during emotional states to prepare the central nervous system for “fight or flight” responses. Both of these monoamines also show circadian rhythms, being highest when awake and active, lower when awake and nonactive, and falling even lower during sleep.
The effect of volume transmission upon a target neuron is more similar to that of a hormone altering the metabolism of its target cells than a traditional neurotransmitter communicating complex information across a synapse. (In fact, monoamines can also act as hormones both inside and outside the brain. As hormones, they are released into capillary beds and then travel through the bloodstream before reaching their targets. However, they can interact with the same receptors whether they travel to their target as neurotransmitters or as hormones).
Regardless, when the extracellular concentrations of serotonin, norepinephrine, and perhaps dopamine, fall below some minimal level, the target neurons in key brain areas are thought to become dysfunctional, resulting in depression.
Types of Monoamine Receptors.
Neurotransmitters can interact with two different classes of receptors on target neurons. Both result in ion flow through the cell membrane that either depolarizes (i.e. excites) or hyperpolarizes (i.e. inhibits) the target neuron. However, the way the two classes of receptors open ion channels and their effects upon target neurons are quite different.
One class, found mainly in synapses, is called an ionotropic receptor. Ionotropic receptors consist of either 4 or 5 protein subunits that come together to form both a low-affinity binding site as well as an ion channel (two functions for the price of one!). In the unbound resting state, the ion channel is closed. However when the neurotransmitter binds the receptor’s binding site, the associated ion channel opens almost instantaneously. Several milliseconds later, when the neurotransmitter falls off the binding site, the ion channel instantly reverts back to its closed state. This short period of receptor binding as well as the quick opening and closing of its ion channels makes ionotropic receptors ideal for detecting brief “bursts” of synaptic neurotransmitter release whose frequency and amount communicate information.
The other type of receptor is called a G-Protein Coupled Receptor (GPCR). GPCRs can be found in synaptic membranes, but also in cell membranes outside of synapses. Unlike the ionotropic receptor, which is a protein complex, the GPCR is a single protein and its ion channel, comprised of 4 protein subunits, is completely separate from the receptor. Because several steps intervene between receptor binding and ion channel opening, it takes longer for a GPCR to open its ion channel, although still pretty quick. But unlike an ionotropic receptor, a single activated GPCR can open many associated ion channels.
Once opened, the GPCR channels also stay open longer. Because of their slower and more prolonged response, GPCRs are not very good for detecting the quick transmission of complex information. On the other hand, GPCRs are very good for modulating the ongoing metabolic activity of target neurons, allowing the target neuron to adjust its metabolism to meet situational demands. Because GPCR’s amplify the neurotransmitter “signal” by each opening multiple ion channels, they are also more responsive to low neurotransmitter concentrations.
Almost all monoamine receptors are GPCRs. Thirteen of the 14 different serotonin receptors are GPCRs and all norepinephrine (n=5) and dopamine receptors (n=5) are GPCRs. These different monoamine receptors are differentially expressed in different parts of the brain presumably to mediate different responses to monoamine input.
However there is still a lot we don’t understand about monoamine contributions to depression. For example, what are the relative contributions of the different monoamines? For example, why are antidepressants that selectively elevate serotonin (SSRIs) approximately equally efficacious to those that selectively elevate norepinephrine and dopamine (NDRIs) ? And why are around 30% of depressed individuals not responsive to any monoamine antidepressants? Inquiring minds want to know!
Evolution of Monoamine Antidepressants
There have been 3 historical goals over the years in trying to improve monoamine antidepressants: 1) making the drugs more effective, 2) making the drugs quicker acting, and 3) reducing the drug’s side effects. In what follows I provide a brief historical overview of the attempts to improve these antidepressants.
First-Generation Drugs. The first-generation drugs: the tricyclic antidepressants (TCAs) and the monoamine oxidase inhibitors (MAOIs) were first introduced in the late 1950’s. Although they work through different mechanisms, they both increase extracellular monoamine concentrations in the brain, with serotonin, norepinephine, and perhaps dopamine thought most important.
In order to understand how these antidepressants work, it is necessary to understand the sequence of events by which monoamine neurotransmitters are synthesized and released. All monoamine neurotransmitters are synthesized from amino acids that come from proteins that you eat. Once digested, the resulting amino acids are absorbed into the blood where they readily cross the blood brain barrier to enter the brain. After entering a neuron cell body, amino acids destined to be converted into neurotransmitters are transported down the axon to the axon terminal. Once inside the axon terminal, enzymes, found only in the terminal, convert the amino acid into the appropriate neurotransmitter (a given neuron synthesizes only one type of monoamine neurotransmitter). Norepinephrine and dopamine are synthesized from an amino acid called tyrosine and serotonin from tryptophan.
Figure 1: Site of action of first generation antideprepressants. Tricyclic Antidepressants (TCA’s) block monoamine reuptake transporters while Monoamine oxidase inhibitors (MAOI’s) block the monoamine oxidase enzyme.
Once synthesized, the monoamine is quickly moved from the axon’s cytoplasm to the inside of a membraneous synaptic vesicle by a vesicular transporter (a protein embedded in the membrane of the vesicle). The synaptic vesicle serves 2 purposes: 1) It is necessary for neurotransmitter release, and 2) It protects the monoamine from monoamine oxidase (MAO), an enzyme lurking in the cytoplasm that would otherwise destroy it (see Figure 1).
To release the monoamine, the membrane of the synaptic vesicle fuses with the presynaptic membrane, and in the process, the monoamine is released into the extracellular fluid (the fluid between the cells). The monoamines then diffuse through the extracellular fluid to bind their receptors on the target neuron in a highly specific lock and key fashion.
Once the neurotransmitter falls off its receptor (usually within a few milliseconds), it is quickly moved back into the axon terminal by a reuptake transporter (a protein embedded in membrane of the axon terminal). Once inside the terminal, the monoamine can be repackaged into a new synaptic vesicle for re-release. This scenario of release, followed by reuptake, followed by release is repeated over and over again (mother nature is a great recycler!). As noted above, synaptic reuptake is much more efficient than non-synaptic reuptake. As a result, the neurotransmitter concentration in the synapse can change dramatically in milliseconds. Removal is further enhanced in serotonin synapses (but not norepinephrine or dopamine synapses) by astrocytes (a type of glial cell) surrounding the synapses that possess serotonin reuptake transporters which augments the removal by the axon terminal. This might suggest that serotonin, unlike norepinephrine and dopamine, can serve as a traditional neurotransmitter as well as a modulatory neurotransmitter, although the implications for depression are not clear.
The tricyclic antidepressants (named for their three-ring structures) increase monoamine concentrations in the extracellular fluid by binding to and blocking reuptake transporters (see figure 2). Without reuptake, continued release causes extracellular monoamine concentrations to increase. However, the first generation antidepressants are not very selective and also bind to other unrelated receptor sites causing many of their undesirable side effects.
The other class of first-generation antidepressants, the MAOIs work by blocking monoamine oxidase (MAO), the enzyme that breaks down monoamines in the cytoplasm (see figure 2). When MAO is blocked, cytoplasmic monoamine concentrations go up, providing more monoamine to be packaged in each synaptic vesicle. The end result is that more monoamine is released by each synaptic vesicle resulting in increased extracellular monoamine.
By some accounts, MAOIs have quicker therapeutic effects and are more effective than the TCAs. Unfortunately, deaths from heart attacks and blood vessel ruptures have occurred when MAOIs are combined with either adrenalin-like drugs or foods that contain an amino acid called tyramine. Consequently, most MAOI’s must be used with extreme caution.
Because of their dietary and drug restrictions, the original MAOIs never got much traction. However, more recently, a more selective MAOI, selegiline, originally used for Parkinson’s disease, has been attracting attention as a depression treatment. Administered as a transdermal patch, selegiline can clear the depression for some patients in days and is not as affected by dietary and drug restrictions. Although compliance is generally good, selegiline can have side effects (insomnia, diarrhea, and sore throat) and may not be effective for all users.
Second-Generation Drugs. These drugs began appearing in the 1970’s and 1980’s and are also referred to as the atypical antidepressants because they work similarly to the tricyclics but with different chemical structures (some of which do not have the three-ring structure). They vary a bit in their modes of action and also have different side-effect profiles making them more suitable for some patients. However, they did not improve on first-generation effectiveness in treating depression.
Third-generation drugs.The most recent drugs, beginning to appear in the 1990’s and early 2000’s account for most of today’s antidepressant use. These drugs fall into three categories 1) Selective Serotonin Reuptake Inhibitors (SSRIs, n = 7 drugs), the Serotonin and Norepinephrine Reuptake Inhibitors (SNRIs, n = 1 drug), and the Norepinephrine and Dopamine Reuptake Inhibitors (NDRIs, n = 3 drugs). All of these drugs are more selective in interacting with brain receptors than previous generation drugs resulting in fewer side effects. But again, they are not more effective in treating depression than the earlier-generation drugs!
So, of the three historical goals in improving monoamine antidepressants, only one has been unequivocally realized: a reduction in side effects. To the credit of the newer drugs, this reduction does result in increased patient compliance.
Selective Serotonin Reuptake Inhibitors.
Mainly because SSRI side-effects are less bothersome for most users, SSRIs are now the first-line drugs for treating depression These drugs are also first-line drugs for anxiety disorders (many depressed individuals also suffer from anxiety), obsessive/compulsive disorder, and can be used off label for other disorders such as ADHD. However, if SSRI’s are ineffective, or if the side-effects are not tolerated, other antidepressants (including the older generation antidepressants) can be tried.
As their name implies, SSRIs are serotonin reuptake inhibitors, although not completely selective, since most also cause some minor inhibition of norepinephrine reuptake. Statistically the 7 different SSRIs all have similar efficacy in treating depression although differences in half-life, potency, enzymatic degradation, regulation of hepatic enzymes, interactions with other medications, and side-effect profiles may dictate their use with particular patients.
SSRI Side Effects. While all SSRIs have fewer side effects than the other monoamine antidepressants, their side effects (like their therapeutic effects) are largely due to serotonin elevations. While not life-threatening, they nonetheless can be bothersome. Fortunately, the side effects typically diminish with continued use.
Following administration, the SSRIs affect serotonin synapses throughout the brain and spinal cord. As pointed out earlier, a bewildering array of different serotonin receptors are differentially expressed in different parts of the brain. While the 1A receptors in the forebrain are thought to be most important for the therapeutic effect, the 2 and 3 receptors located in various brain areas are thought to underly the major side effects (see Figure 3 below).
Figure x. Serotonin is more technically known as 5-hydroxytryptophan (5-HT). This drawing illustrates the effects of raising serotonin concentration on treating depression and producing side effects.
Some of the more problematic SSRI issues are the serotonin syndrome, sexual dysfunction, and when discontinuing, the resulting withdrawal symptoms.
Serotonin Syndrome. The serotonin syndrome arises from an SSRI dosage that is too high or from the additive effect of combining an SSRI with another serotonin-boosting drug. A constellation of unpleasant symptoms, in some cases requiring hospital support, include fever, shivering, high blood pressure, accelerated heart rate and diarrhea. After discontinuing the SSRI, symptoms usually disappear in a day or two.
Sexual dysfunction. Up to 80% of male and female SSRI users suffer some form of sexual dysfunction. Physical issues such as ejaculation problems and vaginal dryness are often accompanied by a loss in sexual desire. One solution is to take another drug to counteract this side-effect such as Viagra (both males and females). A second solution is to switch to another antidepressant that doesn’t cause sexual dysfunction (such as buproprion). While sexual dysfunction often ceases upon stopping the SSRI, for some individuals it persists long after discontinuation.
Discontinuation Syndrome. This problem occurs in many patients who abruptly stop an SSRI. Long-term users, particularly those using SSRIs with shorter half-lives, are most likely to be affected. Like all neuroactive drugs, SSRI use induces pharmacological tolerance. Once established, SSRI discontinuation causes pharmacological withdrawal. Flu-like symptoms, sleep disturbances, gastrointestinal disturbances, dizziness, sensory disturbances, and anxiety/agitation are all possible. The symptoms often last 3 or 4 weeks; but can be reduced by gradually tapering the dosage, although tapering usually extends the withdrawal period. In some cases, the withdrawal symptoms are sufficiently unpleasant that patients initially wishing to quit have chosen instead to stay on their SSRI rather than continuing to experience the withdrawal symptoms.
Popularity of SSRI’s
As mentioned earlier, in addition to being first-line antidepressants, SSRIs are also first-line drugs for obsessive/compulsive disorder, various anxiety disorders and can be used for several other conditions as well. According to the National Center for Health Statistics 12.7% of the U.S. population above the age of 12 reported taking SSRI’s between 2011 and 2014! As a result, SSRIs are “blockbuster drugs” for the pharmaceutical companies that sell them. Estimates are that the SSRIs will generate profits of around 16 billion dollars in 2020. The financial success of these drugs clearly accounts for why there are some many of them and why each pharmaceutical company wants their own.
Effectiveness of SSRI’s
Despite their popularity, about half the patients do not respond to SSRIs initially. Sometimes this can be overcome by upping the dosage, switching to another SSRI or to an earlier generation antidepressant, or by combining several different antidepressants. However even after trying multiple drug strategies, around 30% of patients remain resistant to SSRI’s and other monoamine-related antidepressants.
Published research assessing effectiveness is consistent with SSRIs (and other monoamine-related antidepressants) providing mixed therapeutic value. In fact, an early meta-analysis (combining the results of multiple studies) found that monoamine-related antidepressants (including an SSRI) were no more therapeutic than a placebo for a majority of subjects. Only the most severely depressed had an enhanced response. On the other hand, recent meta-analyses have been more supportive of broader efficacy. But even when beneficial, the average therapeutic benefits are considered modest. Only around 30% of patients treated with SSRIs show a full recovery.
Clearly a significant percentage of patients do benefit to some degree from SSRIs, although I think everyone would agree that we need better drugs. SSRIs do not work more quickly and are not more effective than the older antidepressants, their only advantage is fewer side effects. In the next post, I will discuss some newer drug approaches that hopefully will put us on a path to better antidepressants.
Most experts agree that stress is the critical factor that precipitates major depression. Those prone to depression have greater susceptibility, but presumably everyone has some threshold at which depression would occur.
Over the years there have been at least 5 different hypotheses of what goes wrong in the brain between experiencing stress and the development of depression. However, the explanations are not mutually exclusive, with each hypothesis helping to understand different aspects of the disorder. While each hypothesis has utility, none provides a comprehensive understanding. It is also possible that that major depression is not a single disorder but rather multiple disorders with overlapping symptoms. There is still much we don’t understand.
The Monoamine Hypothesis. The monoamine hypothesis is the oldest hypothesis and derives from the first effective antidepressant drugs that work by boosting monoamine neurotransmitter concentrations in the brain. According to this idea, stress suppresses monoamine neurotransmission which in turn leads to depression.
There are 5 different monoamine neurotransmitters in the brain (serotonin, norepinephrine, epinephrine, dopamine and histamine), however since the monoamine antidepressants target mainly serotonin and norepinephrine, these 2 neurotransmitters have received the most attention. However, there is some evidence that dopamine might play a lesser role.
As illustrated at the end of the previous post, the serotonin and norepinephrine pathways in the brain possess the anatomical and functional characteristics one might expect of systems underlying depression. Although their neuron cell bodies are in the brainstem, their axons terminate throughout the brain, including all the areas involved in depression.
While monoamine neurotransmitters are released across synapses, their role in depression is more likely due to a type of non-synaptic release called “volume transmission.” In volume transmission the monoamine is released into the extracellular fluid outside of synapses where it can affect receptors on multiple nearby neurons. In some cases, the neurotransmitters may be targeted to their receptors by the flow of the interstitial fluid. The receptors that respond are also thought to be largely outside of synapses. Neurotransmitter release by volume transmission is thought to provide a “monoamine bath” in the area of release whose varying concentration modulates or fine tunes local neuronal activity. At the same time, some minimal average concentration is thought necessary to maintain baseline functioning. More about this in the next post.
In the original formulation of this hypothesis, depression was thought to be caused by serotonin and norepinephrine concentrations dropping below some minimal level. However, it was initially puzzling that antidepressants restore brain monoamine concentrations within hours, while recovery takes much longer, typically from three to six weeks.
However, a now well-known principle of neurotransmitter communication is that when neurotransmitter concentrations drop, responding neurons often compensate by upregulating (i.e. increasing) their receptors. This compensation allows continued functioning despite low neurotransmitter concentrations. However, this compensation works only up to a point. Eventually communication breaks down and, in the case of monoamine neurotransmitters, depression results. Consistent with this explanation, the time course to restore normal receptor concentrations with monoamine antidepressants more closely matches the time course for depression recovery. So the thinking is that you must have a proper balance between monoamines and monoamine receptors for normal functioning
However, a problem with the monoamine hypothesis is that monoamine antidepressants do not work for all depressed individuals. Approximately 50% of patients do not respond upon their first try, and after increasing dosage or changing to an alternative antidepressant, about 30% remain non-responsive. Another issue concerns the relative importance of the different monoamines. We’re not exactly sure how the relative concentrations of serotonin, norepinephrine, and dopamine are related to depression onset.
It is perhaps relevant to the monoamine hypothesis that psychedelic drugs that interact with a type of serotonin receptor also seem to provide relief from depression. More on this in a later post.
Hippocampal Neurodegeneration Hypothesis. According to this hypothesis, depression is precipitated by stress causing hippocampal degeneration. Brain scans have demonstrated that the hippocampal volume can shrink by as much as 20-30% in severely depressed individuals. Hippocampal degeneration is then thought to lead to dysfunction in the other brain areas with which it interconnects such as the prefrontal cortex, insula, anterior cingulate cortex, and amygdala. Conversely, successful depression treatment has been observed to increase hippocampal volume and restore functioning in associated areas.
As the model would predict, in stressed rodents with hippocampal degeneration, treatment with a monoamine-related antidepressant increases hippocampal volume. In some respects, the hippocampus neurodegeneration hypothesis might be viewed as an extension of the monoamine hypothesis by specifying the brain area where monoamine dysfunction initiates depressive symptomology.
The hippocampus is certainly best known for its essential role in helping the cerebral cortex to consolidate short-term memories into long-term memories and for its contributions to Alzheimer’s Disease when it degenerates. Hippocampal dysfunction in depression almost certainly also accounts for the memory deficits seen in depressed individuals as well.
One question not answered to everyone’s satisfaction concerns the process that causes the hippocampus to shrink. The hippocampus is thought by many scientists to be the only brain area in adult humans capable of neurogenesis (the birth of new neurons). The argument is that stress turns off hippocampal neurogenesis. When neurogenesis stops, dying neurons are not replaced, and the hippocampus shrinks. While the weight of evidence would seem supportive there are non-supportive findings as well. So whether shrinkage is due to a lack of neurogenesis or some other factor is currently debated.
Circadian Rhythm Abnormality.According to this idea, depression is hypothesized to be caused by the improper functioning of the biological clocks that control our 24-hour circadian rhythms.
All body processes show circadian rhythms with metabolic peaks and troughs at appropriate times of day to optimize body functioning. Although every cell of the body has its own clock, they are all synched to a “master clock” in the hypothalamus of the brain. Surprisingly, human circadian clocks typically have around a 25-hour (rather than 24-hour) periodicity (it can vary a bit from person to person). So, in order for our biological rhythms to be synched to the appropriate time of day, we must set our clocks back about an hour each day. The resetting normally occurs when we wake up each morning by “zeitgebers” (German for time givers), the most important of which is the morning light.
Circadian rhythm abnormalities are often seen in major depression, both in timing and in amplitude. The most obvious expression is in the occurrence of sleep disorders. Sleep issues can occur as amplitude problems where the person gets too little or too much sleep, and/or as temporal problems with difficulties in falling asleep or waking up at the proper time. Physiological variables such as body temperature and hormone secretion often show parallel disturbances. Successful treatment for depression typically reduces these circadian abnormalities.
Interestingly, both serotonin and norepinephrine show pronounced circadian fluctuations in brain concentrations. Both are highest when awake and physically active, drop during wakeful inactivity, drop even further upon entry into sleep, and, in the case of serotonin, drop to virtually zero during Rapid Eye Movement (REM) Sleep.
Several behavioral treatments for depression are consistent with our understanding of these circadian variations in monoamine concentration. For example, sleep deprivation, or even selective REM-sleep deprivation can improve mood in some depressed individuals. These treatments are thought to work by reducing time with low brain monoamine concentrations. Alternatively, boosting day-time monoamine concentrations through exercise can also treat mild cases of depression. Further implicating circadian rhythms, certain gene alleles that code for clock functioning have been linked to higher depression risk.
For people with Seasonal Affective Disorder (SAD), the circadian problems are even more pronounced. During the short day lengths of winter, individuals with this disorder are unable to reset their clocks each day. As a result, circadian clocks begin to “free run”, resulting in physiology losing its synchronization to the time of day. Depression is thought to be precipitated by the resulting dysfunction. While some SAD suffers respond to antidepressants, a more desirable solution is to expose them to an intense bank of lights after awakening each morning to reset their clocks. These individuals are also advised to spend as much time as possible outside during the daylight hours to further insure proper synchronization of their circadian clocks.
Depression Set-Point Abnormality.According to this idea, each individual has a unique set-point for the amount of stress necessary to precipitate depression. Predisposed individuals are thought to have low set-points while depression-resistant individuals have higher set points. For reasons that aren’t altogether clear, electrically stimulating the brain is thought to raise the set-point and, in so doing, treat depression. Several different stimulation methods have some degree of effectiveness. For a variety of reasons, these methods are mainly “last-resort” therapies when other approaches don’t work. These procedures are sometimes used for other brain disorders as well.
Electroconvulsive shock therapy is the most effective non-drug treatment available for patients unresponsive to antidepressants or psychotherapy. Electrodes placed on the skull to affect both hemispheres, or just one hemisphere, transmit high-voltage, low-amperage electricity through the cortex, resulting in an epileptic-like seizure. The procedure is performed in a hospital settings under sedation with individuals normally receiving 8-12 treatments over a period of 3 or so weeks. Some patients subsequently undergo maintenance treatments around one month apart to prevent relapse.
Memory issues and headaches can occur immediately after treatment, but typically clear up with time. While the treatment may sound terrible (particularly if you saw the movie “One Flew Over the Cuckoo’s Nest”), this treatment has been highly refined over the years and is considered safe. Research indicates that this treatment has an overall success rate of 90% in some studies, making it more effective than monoamine antidepressants. In some cases, electroconvulsive shock has also provided lifesaving relief from suicidal ideology.
Repetitive transcranial magnetic stimulation (rTMS) is a less invasive alternative to electroshock therapy, particularly for patients with less treatment-resistant depression and is most often used to augment antidepressant therapy and/or psychotherapy. Neurons in the prefrontal lobe are repeatedly stimulated by a powerful alternating magnetic field from magnets placed outside the skull. Like electroshock therapy, the treatment can be either bilateral or unilateral. The patient typically receives treatments for 5 days per week over a period of 4-6 weeks. Unlike electroshock therapy this procedure can be performed in a doctor’s office or clinic and does not cause epileptic seizures or require sedation. When used alone, it is less effective than electroshock therapy but approximately as effective as antidepressant drugs.
Yet another way of stimulating the brain is by stimulating the vagus nerve, a cranial nerve that services much of the body. A stimulating device is surgically implanted under the skin of the chest with a wire connecting to the left vagus nerve where it passes through the neck on its way to the brain. By repeatedly stimulating the vagus nerve, various parts of the brain are also stimulated. However, it may take several months of treatment to see effects on depression and is less consistently effective than either electroshock or transcranial magnetic stimulation.
Although more commonly used for Parkinson’s Disease, deep-brain stimulation (DBS) has been used to treat depression and is reasonably well tolerated. Electrodes are surgically implanted into the brain to deliver electrical stimulation directly to brain tissue. A number of brain sites have resulted in therapeutic effects. Despite discrete electrode placements, this approach does not allow for inferences about therapeutic brain sites since the stimulation quickly spreads to areas outside the stimulation site. Because of its invasiveness, this treatment is rarely used for depression.
Glutamate hypothesis. According to this hypothesis, the overrelease of glutamic acid, a brain neurotransmitter, causes the brain dysfunctions that underly depression. This hypothesis is based upon the finding that ketamine, an anesthetic drug that acts as a glutamic acid antagonist, has antidepressant properties.
Glutamic Acid (GA), also referred to as glutamate, is the major excitatory neurotransmitter of the brain and is central to the functioning of all the forebrain structures implicated in depression. Paradoxically, too much GA neurotransmission causes “excitotoxicity” (which poisons the brain). GA neurons are normally kept in check by neurons that release Gamma Amino Butyric Acid (GABA), the principle inhibitory neurotransmitter in the brain. Abnormal Levels of both GA and GABA have been observed both in depressed humans and in animal models of depression.
Support for the importance of GA neurons was first provided in 2010. In the initial study, depressed individuals, previously determined to be unresponsive to the therapeutic effects of monoamine antidepressants, received a single subanesthetic injection of ketamine. 80% showed symptomatic relief! Moreover, the therapeutic effects occurred within hours (rather than the weeks typical for the monoamine antidepressants). Unfortunately, the effects typically wear off within a week or so; however, some patients have had successful maintenance therapy for over a year, with 2 to 7-day dosing intervals.
Despite the seeming success of ketamine, it is not an ideal antidepressant. Because it is a DEA-controlled drug with short-term debilitating effects and potentially addictive properties, self-administration is illegal. The treatment by a physician is also costly, and, since it is not FDA-approved for this purpose, insurance typically does not cover costs. Currently, it is used mainly for patients unresponsive to monoamine antidepressants.
Although a new form of ketamine (esketamine) recently received FDA approval for treating depression, making it eligible for insurance coverage, it still must be administered by a physician and possesses the same side effects. A drug that provides ketamine’s benefits without its downsides, would certainly be highly desirable. There are currently attempts to develop such drugs. More about that in a later post.
To complicate matters further, GA neurons and monoamine neurons are reciprocally interconnected. The GA neurons of forebrain areas that contribute to depression are modulated by extracellular monoamine concentrations, while GA synaptic input into serotonin, norepinephrine, and dopamine neurons undoubtedly influences monoamine secretion as well. Ketamine’s antidepressant action also restores normal concentrations of brain monoamines.
Still a lot of pieces of the depression puzzle that don’t yet fit together!
Approximately 10% of males and 25% of females will experience a major depressive episode sometime during their lifetime. It is the 3rd most disabling disease in the world accounting for 70% of the psychiatric hospitalizations and 40% of the suicides. In addition to feeling sad, depressed individuals can experience a wide range of other symptoms. Chronic anxiety, emptiness, hopelessness, and worthlessness are common, as are eating and sleeping disorders. Activities that were previously pleasurable (including sex) often seem unrewarding. Low energy levels, slowness of movement, and chronic fatigue are also common as are physical symptoms that don’t respond well to treatment (such as chronic pain or digestive issues). And in some cases, cognitive and memory impairments bear a remarkable resemblance to the early stages of Alzheimer’s Disease. Depression also exacerbates other illnesses and reduces life expectancy.
In this post, I provide a brief overview of the neuroanatomy of depression. In a second post, I review some hypotheses of the causes of the brain dysfunctions underlying depression. In a third post, I describe the monoamine-related antidepressant drugs and their evolution over time. And in yet other posts, I discuss new classes of antidepressants that provide relief for many individuals that do not respond to traditional monamine antidepressants.
Where in the brain is depression located.
In what follows, I briefly present some brain structures known to be dysfunctional in depression and the specific roles they are thought to play . I also try to give you a feel for where these structures are located in the brain. For cortical structures on the surface of the brain, I use a graphic showing the left hemisphere and for structures buried inside the brain, I use a midline section.
Given the emotional, cognitive, and motor symptoms of depression, it should not be surprising that many brain areas are involved. Since the brain areas all communicate with each other, it is not clear if malfunctioning in one area is primary, or secondary to malfunctioning in another. In fact most depression symptoms likely arise from the interaction of multiple brain areas.
Cortex. Depression can be viewed as an altered state of consciousness, so it should not be surprising that the conscious part of our brain, the cerebral cortex is involved. The cortex is the convoluted grey matter providing the forebrain’s outer covering (see Figure 1, click on figures to enlarge them if text is too small).
Figure 1: Sensory Cortex is green, Motor Cortex is red, with the tan remainder being Association Cortex
Different parts of the cortex contribute to consciousness in different ways. Sensory cortex (green color) decodes the different types of sensory input and makes the information available to consciousness, while motor cortex (red color) directs motor output for conscious behavior. However, most of the cortex does not have well defined sensory or motor functions and is called association cortex (tan color).
One important function of association cortex is to encode long-term memories so they can be made available to consciousness. (Memories stored outside the cortex are not available to our conscious minds). Long-term cortical memory storage occurs through the creation of new synapses or the modification of existing ones. Conscious memory is also organized according to principles that facilitate recall. For example memories are stored near the sensory cortex that predominates in that memory; visual memories are stored near primary visual cortex , auditory memories near primary auditory cortex and so forth. As they are encoded, memories are also “time-stamped,” and “location-stamped” so you can typically identify when and where the memory occurred. And finally memories are also “cross-indexed” with other relevant memories providing multiple paths for retrieval. These processes are all impaired to some extent in depressed individuals.
The other important function of association cortex is to provide executive control over the planning and decision-making processes of the cortex. This role is carried out primarily by the prefrontal cortex which can attend to relevant relevant sensory input, compare that input to stored memories to decide what to do, and then direct motor cortex to perform whatever actions best achieve the desired goal. Basically the prefrontal cortex makes decisions about what you should be doing and when you should be doing it. These processes are also clearly impaired in depressed individuals. If we were to designate a part of the human brain as the “seat of consciousness” it would be the prefrontal cortex.
Figure 2: The prefrontal cortex is the executive part of association cortex that is central to the conscious planning and decision making processes of the cortex
When a person is depressed, the prefrontal cortex exhibits a dysfunctional metabolic state called “hypofrontality” (also present in schizophrenia and other psychiatric disorders). Hypofrontality is accompanied by decreases in blood flow, neural activity, and neuronal metabolism. In some cases, there is even evidence of degeneration. As the final common pathway for conscious thought and behavior, the malfunctioning prefrontal cortex is clearly central to many depression symptoms.
As might be expected, after depression recovery, prefrontal functioning typically returns to a more normal state. Further implicating the prefrontal cortex , a depression treatment (repetitive transcranial magnetic stimulation), targets the prefrontal cortex. An even more effective, but less targeted treatment, electroconvulsive shock, is also thought to work by “jump starting” the prefrontal cortex. And finally consciousness-altering drugs, such as ketamine and other psychedelic hallucinogens, might also exert their therapeutic effects by re-activating prefrontal cortex activity. More about these treatments in a later post.
Figure 2. The anterior cingulate cortex encodes memories for the expectation of reward based upon past experience.
At the same time, proper functioning of the prefrontal cortex also depends upon appropriate input from other cortical structures. Two structures that have received considerable attention are the anterior cingulate cortex and the insula. The anterior cingulate cortex is buried deep in the longitudinal fissure (the deep groove in the top of the brain that separates the two hemispheres) right behind the prefrontal cortex. Among other things, the anterior cingulate cortex encodes the expectation of reward in different situations based upon experience. Its malfunctioning is thought to contribute to the depressed patient’s lack of interest in activities that were previously enjoyable. The insula, hidden inside the lateral fissure (the deep groove running horizontally on the side of the cortex) is involved in the immediate self-awareness of one’s feelings and behavior and helps filter out disruptive negative stimulation. Insula malfunctioning is thought to contribute to the increased salience of negative stimuli and the increased rumination of negative thoughts.
Figure 4: The insula or insular cortex is buried inside the lateral fissure, the deep groove that forms the “thumb of the boxing glove.” The insula can not been seen on the outside of the brain
The limbic system and basal ganglia, two subcortical forebrain systems, are massively interconnected with the cortex, and play important roles in supporting cortical functioning. The hippocampus is essential in helping the cortex transform short-term memory (transient electrical events) into long-term memory (structural changes in cortical synapses). Without proper hippocampal functioning a person is unable to encoded new long-term memories. The hippocampal dysfunction during depression is thought to account for the Alzheimer’s-like memory deficits often seen in severely depressed individuals. In fact, one school of thought is that hippocampal degeneration may be the underlying cause of depression.
Figure 5. The limbic system and the basal ganglia are two subcortical forebrain systems essential to proper cortical functioning.
The other important limbic system structure, the amygdala, supplies emotional information to the cortex. Through experience, the amygdala assigns positive or negative value to environmental stimuli as well as to the behaviors that allow you to interact with these stimuli. This information is subsequently incorporated into long-term memories stored in the cortex. The amygdala dysfunction seen in depression is thought to contribute to the inability to experience reward as well as the enhanced negative emotional expression prevalent in depression.
The basal ganglia are a series of interconnected subcortical structures deep inside the forebrain that help the cortex plan and execute motor movements. The motor problems of both Parkinson’s and Huntington’s Diseases are caused by the selective deaths of basal ganglia neurons. While the issues appear less serious in depression, basal ganglia dysfunction is thought to contribute to the low energy levels and slowness of movement seen in Depression.
Figure 6: The Raphe Nuclei are a series of brainstem structures whose neurons send their serotonin-releasing axons to virtually every location in the brain and spinal cord.
And finally, two brainstem structures that contribute are the Raphe Nuclei and Locus Coeruleus. Both are relatively small structures located in the dorsal pons of the hindbrain that house the cell bodies of neurons that release monoamine neurotransmitters. The Raphe nuclei contain the cell bodies of the brain’s serotonin-releasing neurons while the locus coeruleus contains the cell bodies of the brain’s norepinephrine-releasing neurons. In both cases, their long axons travel to virtually all areas of the brain and spinal cord where they modulate the activity of the neurons they innervate. All of the brain areas involved in depression receive input.
Figure 7: The locus coeruleus is a brainstem structure whose neurons send their norepinephrine-releasing axons to virtually every location in the brain and spinal cord
While neurotransmitter release by both of these systems exhibit circadian rhythms, release also increases under specific circumstances. Serotonin release increases immediately before executing body movements and are thought to prepare the different levels of the nervous system to efficiently carry out movements. Norepinephrine, on the other hand, is the neurotransmitter of “sympathetic arousal” and helps to prepare the nervous system to deal with emotion-provoking situations which also often require movement (in extreme situations: “fight or flight”).
The serotonin and norepinephrine systems target many of the same brain areas and have similar patterns of circadian release. Both also decrease their activity during depression. Consistent with the role of these systems in depression, traditional monoamine-related antidepressants are therapeutic by boosting serotonin and/or norepinephrine concentrations in the brain. However, monamine-related antidepressants don’t provide therapeutic relief for all depressed individuals, and the exact role of serotonin, norepinephrine, and perhaps dopamine, in depression is incompletely understood.
We’re not exactly sure how all these brain dysfunctions come to be. Is there an initial issue that causes all the other problems or do all the problems arise independently? In the next post, I will present some of the ideas scientists have had over the years.
There is no “cure” for drug addiction, however there are drugs currently being used, or being developed, to help addicts cope and to help them quit. A comprehensive listing is beyond the scope of this post. Here, I focus more on the strategies than on the drugs themselves.
When employed, results are often mixed, working better for some addicts than others. Even when there is some effectiveness, compliance is often a problem. Since some of the treatment drugs are themselves addictive, some people are philosophically opposed to substituting one addictive drug for another. Some critics also argue that treatment drugs are just temporary “crutches.” Many treatment strategies are also complicated by addicts being addicted to more than one drug. Nonetheless for some individuals, these treatment drugs clearly improve the addict’s situation.
I am not a clinician so please consult a more comprehensive source or a clinical professional for information/advice about specific treatments. It’s very sad that around 40 to 60% of recovering addicts relapse within 1 year. At the end of this post, I provide links to resources which might be helpful.
Different ways that a drug can interact with a receptor.
There are variety of ways a therapeutic drug can interact with brain receptors to produce its effect. A drug can be a full agonist, partial agonist, neutral agonist (also called a receptor blocker) or an inverse agonist for the brain receptor that both it and the addictive drug utilizes. A full agonist is capable of causing a receptor system to produce its maximal response, with the others have progressively less capability, with the inverse agonist actually producing the opposite effect. All of these classes of drugs have been used in the treatment of drug addicts. Figure 1 illustrates the differences in receptor responsiveness to these different classes of drugs.
Fig 1. Idealized dose response curves of an agonist, partial agonist, neutral antagonist, and inverse agonist. (https://en.wikipedia.org/wiki/Inverse_agonist
Full Agonist substitution.
Agonist substitution basically involves switching to a different, but equally efficacious, drug and/or to a different method of administration that is less harmful. The systematic use of this strategy began with heroin addicts substituting a synthetic opiate called methadone, another full opiate agonist. This strategy has since been applied to other drugs of abuse (e.g. nicotine gums for cigarettes, oral cannabis for smoked marijuana, amphetamines for cocaine). This strategy, when employed, attempts to achieve a number of goals.
One goal of this strategy is that the substitute drug should enter the brain more slowly so that it begins binding receptors more slowly while still satisfying the addict’s drug need. Slower initial binding causes less of a “rush” and less euphoria, allowing the addict to function more normally. This outcome can be accomplished in a variety of ways: by using a less lipophilic (fat-like) version of the drug which slows passage across the blood/brain barrier, by having the drug taken orally or by skin patch which causes slower entry into the blood and ultimately into the brain (versus intravenous, smoked, vaped, or snorted).
A second goal is that the substitute drug possesses a longer half-life to even out the addict’s drug response. For example, heroin’s relatively short half-life results in the addict’s entry into unpleasant withdrawal several times a day as the heroin begins to wear off. With the much longer half-life of methadone, these ups and downs are eliminated. In addition, a longer half-life also reduces the intensity of withdrawal should the addict miss a dose. Amphetamine (whose effects are virtually indistinguishable from cocaine) is sometimes substituted for cocaine, in part, for it’s longer half life. A longer period of effectiveness can also be achieved through timed release formulations.
A third goal is shifting to a safer method of administration. In the case of heroin users, that involves shifting from intravenous (I.V.) administration, which is fraught with disease hazard from dirty needles, to a much safer oral methadone administration. Oral amphetamine is similarly viewed as safer than administering cocaine by either I.V. administration, snorting or vaping. Nicotine gum or skin patch is similarly viewed as less harmful than smoking.
And finally, from an economic/societal perspective, providing addicts access to legal drugs (such as methadone) is substantially cheaper than incarcerating them for using illicit ones. While this approach can also be better for the addict’s wellbeing, it doesn’t necessarily turn the addict into well-functioning member of society. However, it can reduce the addicts’ use of illegal drugs as well as criminal behavior to support their habit.
Partial Agonist substitution.
This strategy involves using an alternative drug that binds to the same receptor as the addictive drug, but is only capable of producing a partial response. The addict’s drug need is hopefully satisfied while producing a much milder high. At the same time, should the subject relapse, the substitute also serves as a receptor blocker to keep the original drug from exerting its more powerful effect. Some examples of this strategy are buprenorphine (e.g. Subutex ) for opiate addicts and varencline (Chantix and Champix) for nicotine addicts (used in smoking cessation).
Receptor Blockers (Neutral Agonists).
The idea here is that if the drug is no longer rewarding, the user will not use it and (hopefully) eventually lose interest in taking it. One way of keeping a drug from being rewarding is to block its ability to bind to brain receptors.
A receptor blocker works by binding the same receptor site as the addictive drug but, unlike the addictive drug, produces no effect on its own. However, by binding the receptor site, ideally with higher affinity than the drug, the blocker prevents the addictive drug from binding the receptor. Probably the best-known examples are naloxone (e.g. Narcan ) and naltrexone (e.g. ReVia and Vivitrol) used to block the rewarding effects of heroin, fentanyl, and prescription opiates (and also can be lifesaving in treating opiate overdose). One downside to the regular use of receptor blockers is that it causes the blocked receptors to upregulate (i.e. increase in numbers). If the addict should discontinue taking the receptor blocker and perhaps a few days later begin taking the addictive drug again, the increased number of induced receptors greatly enhances the likelihood of overdose.
Inverse Agonists.
Inverse agonism can occur if a receptor system possesses some degree of spontaneous activity in the absence of agonist drug binding. The inverse agonist can then suppress the spontaneous activity to produce its negative effect. Therapeutic drugs with this capability would probably be used more for their receptor blocking properties than for their inverse agonism (which ideally would be of small magnitude).
Rimonabant (Acomplia, Zimulti), a cannabis CB-1-receptor inverse agonist, provides a cautionary tale. This drug could, of course, be used to block the rewarding effects of marijuana. However, rimonabant was introduced in Europe in 2006 as a diet pill (by blocking food reward) and had off label use as an aid in smoking cessation (by blocking nicotine reward). Shortly thereafter, in 2008, rimonabant had to be withdrawn from the European market (and also was not approved for use in the U.S.) because its use was associated with an increased incidence of psychiatric problems including depression and suicide.
Some now think endogenous cannabinoids working through CB-1 receptors may help many forms of reward turn on the dopamine reward circuitry accounting for rimonabant’s therapeutic uses described above. However, rimonabant’s side effects not only disqualify it as a therapeutic drug, they also provide serious concerns for other therapeutic strategies designed to suppress the general capacity to experience reward.
Aversion Therapy Drugs.
The idea here is that if the use of a particular drug is made aversive, the addict will be disinclined to use it. Disulfram (Antabuse), a drug developed to treat alcoholics, makes alcohol consumption aversive by blocking aldehyde dehydrogenase, the enzyme that eliminates aldehyde buildup following alcohol consumption. This drug normally has little effect on its own. However when the alcoholic takes a drink, the resulting toxicity causes flushing, nausea, vomiting and anxiety. Needless to say, disulfram compliance can be a problem.
Drug Vaccines.
Another strategy that might be available in the near future is using vaccines against specific drugs. The antibodies stimulated by the vaccine would attach to the drug preventing it from crossing the blood/brain barrier. Without access to the brain, the drug would not be able produce its rewarding effects. However, you have to “trick” the immune system to get it to produce the required antibodies.
The problem is that most neuroactive drugs (e.g. cocaine, heroin, nicotine, etc.) have to be very small lipid molecules in order to slip through the blood/brain barrier. However, their small size normally prevents detection by the immune system. To make these small drugs recognizable, the drug must first be modified and then attached to a much larger carrier protein. If done correctly, such a drug/protein complex can then be used to make a vaccine that will stimulate antibodies against the drug. Should a vaccinated addict take the drug, the drug antibodies can then attach to the drug and prevent it from exerting its effects. ( A video by the NIH describes the process in more detail.)
While the technique works in principle, the problem so far has been in getting the human immune system to produce sufficient antibodies, or sufficiently active antibodies, to provide meaningful protection. However, vaccine developers haven’t given up, and vaccines for many drugs of abuse are currently in development (e.g. cocaine, nicotine, methamphetamine, fentanyl, fentanyl analogs, heroin, and oxycodone). A downside is that vaccination works only for the drug you have been vaccinated against. Other drugs could still be abused for their rewarding value.
Detoxification and Rehabilitation.
The drug strategies mentioned are used both in easing the addict’s ongoing problems and in trying to quit. All treatment strategies are more likely to work if the addict is strongly committed to the treatment. However, as noted earlier, I am not a clinician, so for a broader overview of drug rehabilitation, I refer you to an excellent on-line document by the Substance Abuse and Mental Health Administration (SAMHSA) entitled “What is Substance Abuse: A Treatment Book for Families?”
People seeking treatment should certainly research the possibilities first. If you’re concerned about alcoholism treatment, I also recommend a relevant article published in the New York Times.
In two previous posts, I addressed issues in defining addiction, how the brain’s reward circuitry is involved, and suggested that addiction is caused by a highly maladaptive form of learning in unconscious parts of the brain that are highly resistant to conscious influences. In this post, I briefly address the role of genes and environment in addiction.
How do genes affect addiction?
While genes clearly contribute to addiction, they do not cause addiction! The way genes contribute is by providing a predisposition that may or may not be expressed depending upon environmental circumstances.
Addiction does sometimes run in families and many human behavioral genetic studies have used family data to estimate the heritability of addiction to various drugs. While estimates vary, all find a heritability greater than zero with the average of all the studies being around 0.5. A heritability of 0.5 would mean that, on average, 50% of the differences seen among individuals are caused by underlying genetic differences and 50%, by underlying environmental differences. However, heritability is a population statistic that tells you nothing about specific individuals. In addition, single genes with large contributions to addiction liability have not been discovered. The genetics likely involve many genes interacting in complex ways that may be somewhat different from one person to the next.
Clearly some individuals appear more predisposed than others and require less drug exposure, although we have little understanding of the genes that might be involved. There is some evidence that the sensitivity of the brain reward circuitry and functioning of the frontal lobe may be different in some predisposed individuals. In other cases, it may be as simple as differences in the ability to be affected the drug. For example, when I was approaching adulthood in Texas, we thought it “manly” to be able to “hold your liquor” and admired peers who could drink a lot with minimal outward effects (not sure what this says about me). However, we now know that such individuals are significantly more likely to become alcoholics. Again, this trait doesn’t dictate that you will become an alcoholic, but it does increase the likelihood. This principle likely applies to other addictive drugs as well. People less responsive to an addictive drug’s incapacitating effects will likely consume more of it, and more regularly, thereby increasing the risk of addiction.
At the same time, some genetic predispositions may be from genes promoting behaviors that, for whatever reason, simply increase the likelihood of using an addictive drug. Although using a drug doesn’t necessarily result in addiction, it does statistically increase the likelihood. In the following, I provide some examples.
A genetic predisposition to alcoholism might be whether you like the taste. For example, one genetic strain of mice (C57BL/6J) prefers water adulterated with alcohol while another (DBA/2J) avoids it altogether. Similar preferences in humans could promote alcohol use and, in some of those who abuse it, result in addiction. Addiction to other drugs might be affected by similar ‘likes”
In addition, many psychiatric conditions are associated with increased addiction risk. The increased likelihood seen among untreated individuals with ADHD is thought to be related to their higher impulsiveness and lower self-control. In the case of individuals suffering from schizophrenia, bipolar disorder, anxiety, or depression, the effects of the addictive drug sometimes overlap those of the therapeutic drugs used to treat these disorders (there is no clear boundary between addictive and therapeutic drugs!). While the initial motivation may be self-medication for symptomatic relief, with abuse, addiction is a potential outcome.
Conversely, if you avoid a drug, addiction to it is impossible. In the earlier example, an innate dislike for the taste of alcohol should provide protection against alcoholism. Additionally, there are individuals possessing a defective enzyme that allows the toxic buildup of aldehyde after drinking alcohol. These individuals experience extremely unpleasant symptoms, almost never drink alcohol after their first experience, and are at virtually no risk for developing alcoholism. A defective enzyme for nicotine degradation similarly reduces the risk for becoming an addicted smoker. At the same time these protections would not affect risk for other drugs.
So, genes are clearly involved in addiction, but the paths by which they exert their effects are often indirect and variable from person to person.
How do environmental factors contribute?
Although the focus here has been on genetic factors, the environment is approximately equally important. As with genes, the environment’s contribution is complicated and may vary from addict to addict. Like genes, environmental factors that promote drug use, or even drug access, are associated with a higher risk. Peer pressure, low educational opportunities, scarce job opportunities, low recreational opportunities, and environments in which drug dealers are role models, are all associated with a higher risk. Additionally, stress is definitely a factor in precipitating drug use. While I have focused on biological factors in my blogs on addiction, any comprehensive understanding of addiction must take environmental factors into consideration as well.
To learn more.
Advocat, Comaty & Julien (2019). Chapter 4: Epidemiology and neurobiology of addiction. In Julien’s Primer of Drug Action. Thirteenth Edition. Worth Publishers. 267-295. (A good textbook that I last used in my teaching. Many references to primary literature at the end of this chapter)
Charles P. O’Brian. Drug use disorders and addiction. Chapter 24 in Goodman and Gilman’s The pharmacological basis of therapeutics. McGraw-Hill Publishers. (written for medical professionals so fairly technical)









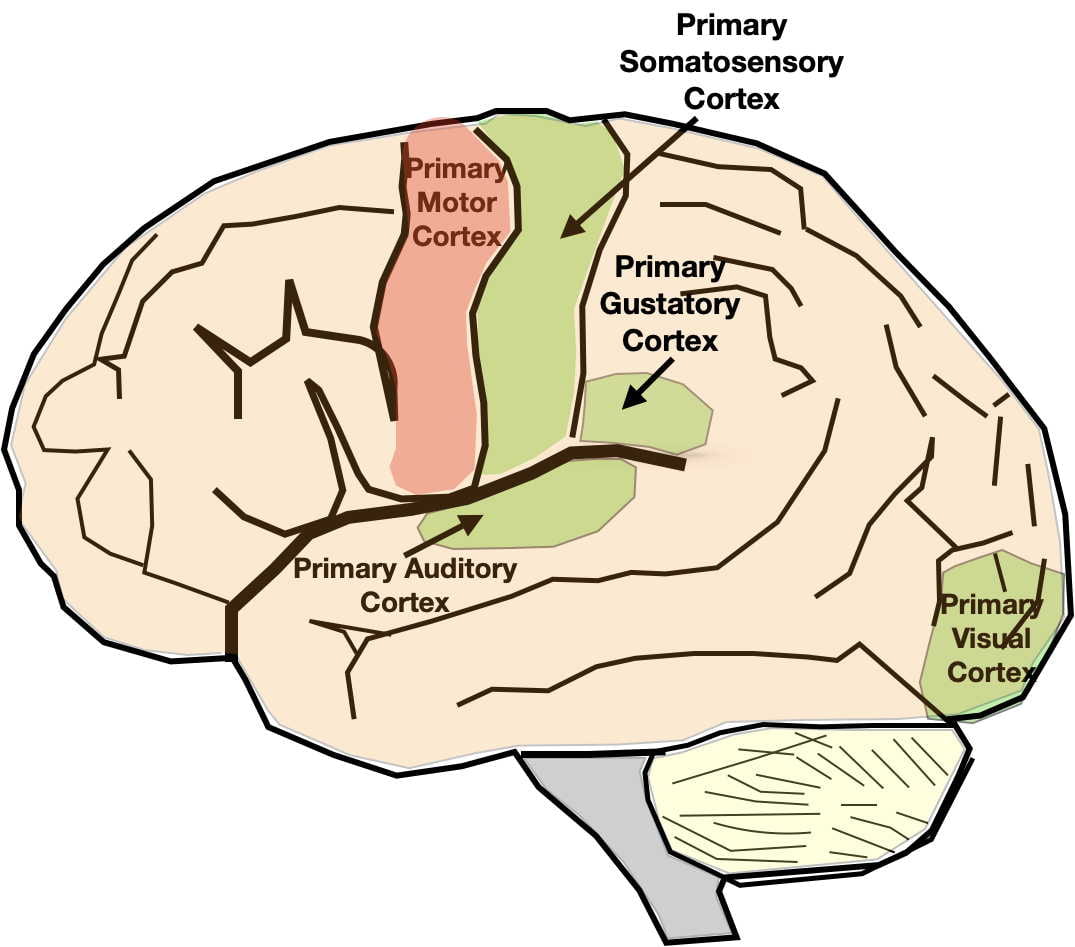

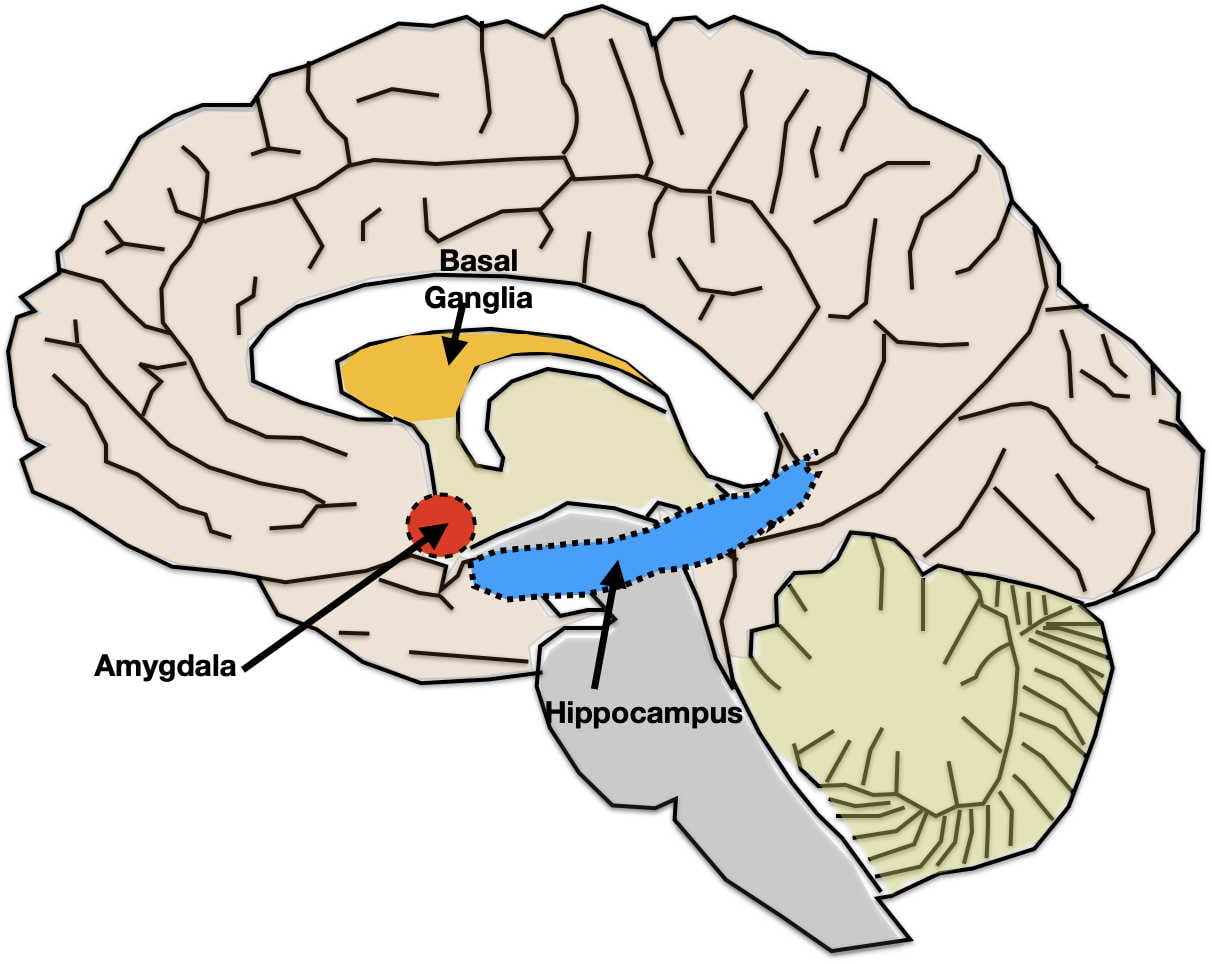
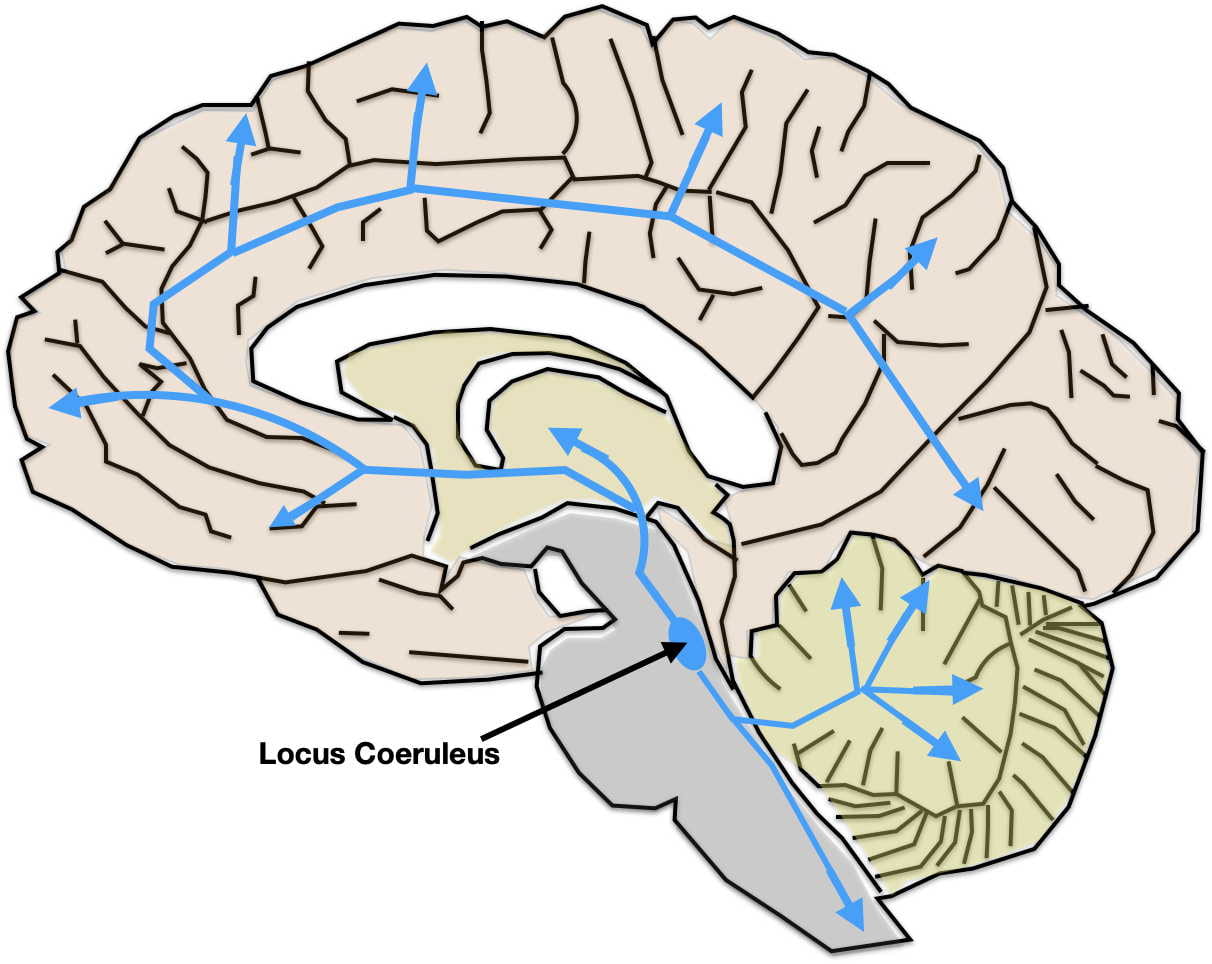


{kind=link}